

Versión 4
INTRODUCCIÓN
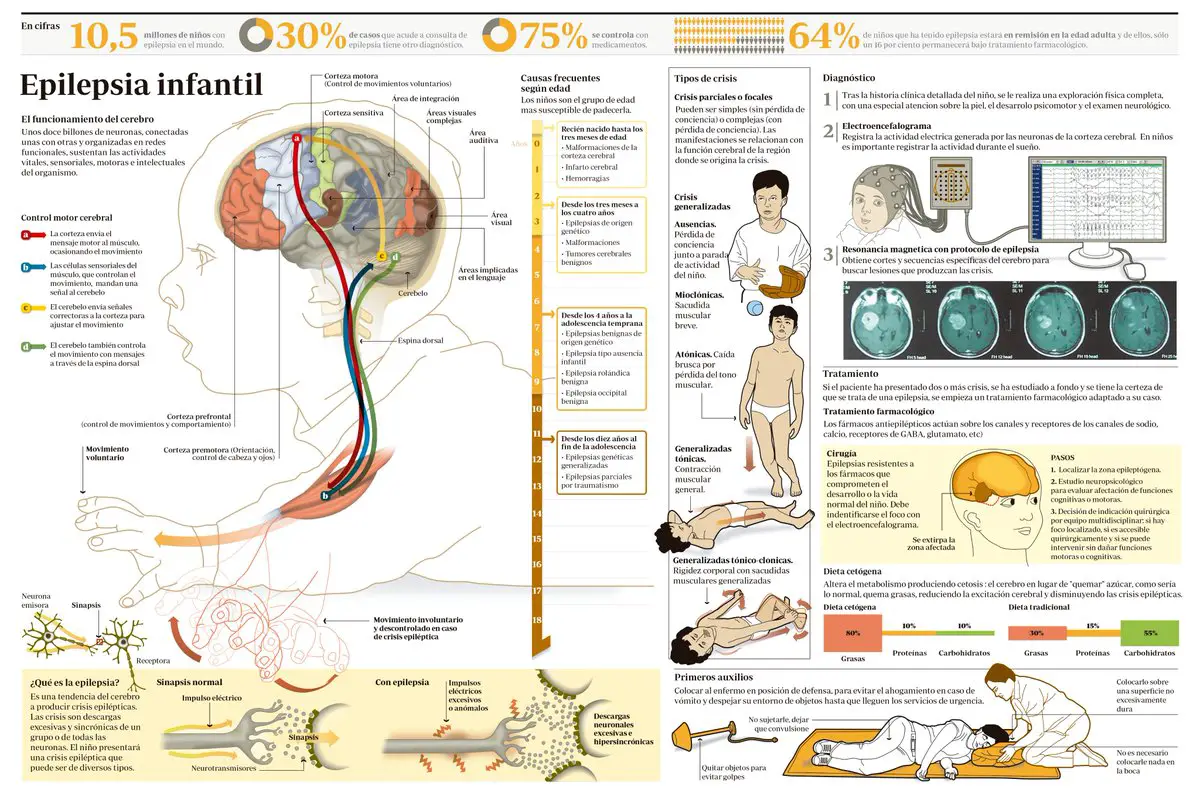
La epilepsia representa uno de los trastornos más comúnmente observados en la práctica clínica en el campo de la neurología, y muy especialmente en el de la neuropediatría. La prevalencia general de la epilepsia infantil es del 0,4-0,8%, lo que supone una incidencia de 20-70/100.000/año. Esta cifra se eleva hasta el 2-10% en la población infantil. La evolución natural de la epilepsia en la infancia estará determinada por el tipo de síndrome epiléptico (Kwan y Sander, 2004). El 20-30% remitirá de manera espontánea, otro 20-30% lo hará con tratamiento y el 30-40% serán epilepsias refractarias.
ANTECEDENTES HISTÓRICOS
La epilepsia ha estado presente en todas las sociedades desde la antigüedad más remota, en occidente está documentada desde el 1760 a.C. (Código de Hammurabi). En Grecia y Roma se denominó morbo sacro («enfermedad sagrada»). Hipócrates (siglo IV a.C) fue el primero en considerar que tenía su origen en el cerebro y no en posesiones de deidades o castigos divinos. En su tratado Sobre la enfermedad sagrada, afirmaba que esta enfermedad tenía la misma naturaleza que el resto de las enfermedades, y calificaba como ignorantes a aquellos que le atribuían una causa divina.
Durante la Edad Media, las teorías naturalistas de los médicos griegos perdieron influencia para dar paso a creencias demoníacas (San Isidoro de Sevilla popularizó en el siglo VII el término lunaticus. A partir del Siglo de las Luces (siglo XVIII), se inicia el declive de las hipótesis demoníacas y se empiezan a utilizar términos como «telele» o «tembleque». En el siglo XIX, algunos autores mantenían, desde una óptica moralista (moral católica), la hipótesis de algunas prácticas de «degeneración moral», como el onanismo (masturbación), en el origen de la epilepsia. Esta concepción perduró hasta el siglo XX, acarreando terribles consecuencias en forma de tratamientos supuestamente antiepilépticos como la castración, la ooferoctonía y la clitoridectomía.
«La evolución histórica de la epilepsia ha sido descrita como 4.000 años de ignorancia, superstición y estigma, seguidos por otros 100 años de conocimiento, superstición y estigma».
Kale (1997)
DEFINICIONES Y CONCEPTOS BÁSICOS ACTUALIZADOS EN EPILEPSIA
En el año 2010, la Liga Internacional contra la Epilepsia (ILAE) estableció y actualizo los principales conceptos en epilepsia infantil (Berg et al., 2010):
- Epilepsia: trastorno cerebral caracterizado por una predisposición duradera a generar crisis epilépticas y por las consecuencias neurobiológicas, cognitivas, psicológicas y sociales de esta condición.
- Crisis epiléptica: ocurrencia transitoria de signos y/o síntomas derivados de una actividad anormal excesiva o síncrona de la actividad neuronal.
- Una única crisis puede ser suficiente para establecer el diagnóstico con un electroencefalograma (EEG) acorde (en el clásico criterio ya superado se precisaban 2 crisis o más).
- Síndrome epiléptico: grupo de signos y síntomas que configuran un trastorno epiléptico único. Dos grandes grupos que engloban diferentes entidades concretas, pero que tienen en común el curso clínico:
- Encefalopatía epiléptica o cuadro epiléptico en el que las crisis contribuyen a un deterioro progresivo de las funciones cerebrales.
- Síndrome epiléptico benigno o síndrome epiléptico caracterizado por crisis fácilmente tratables que requieren o no medicación y pueden remitir espontáneamente con el paso del tiempo.
- Epilepsia refractaria: persistencia de crisis epilépticas diagnosticadas con certeza, que afectan a la calidad de vida, tras haber tomado dos fármacos en monoterapia y una asociación de dos fármacos de primera línea, apropiados al tipo de crisis y síndrome epiléptico, a las dosis terapéuticas máximas toleradas, con cumplimiento adecuado, durante un período de 2 años, que puede ser acortado en períodos de gravedad (Go?mez-Alonso y Bellas-Lamas, 2011).
«La definición de EPILEPSIA REFRACTARIA ha llevado a confusión en el pasado hasta que de forma internacional se ha llegado a un consenso aceptando la E. REFRACTARIA como un tipo de epilepsia que es resistente al tratamiento farmacológico. La explicación que se da en el párrafo que Vd. alude es más completa porque contempla todos o casi todos los parámetros a tener en cuenta como la utilización en el tto. de 2 fármacos , dosis utilizadas, período de 2 años mantenido de tto….etc…No obstante, le sugiero que entienda como Epilepsia Refractaria la definición más simple que la he señalado en negrita y que es la aceptada a nivel internacional por la ILAE (International League Against Epilepsy)». Dra. del Cerro
CLASIFICACIÓN DE LAS CRISIS EPILÉPTICAS
La primera condicio?n que hay que abordar para definir una crisis epile?ptica se refiere a la afectacio?n de la conciencia durante su transcurso. En funcio?n de este aspecto se diferencian dos tipos de crisis:
- Crisis parciales o focales: la conciencia no se pierde total y absolutamente desde el inicio de la crisis. Pueden ser:
- Parciales simples: conciencia totalmente conservada.
- Parciales complejas: conciencia afectada.
- Parciales secundariamente generalizadas: crisis parcial que posteriormente se generaliza.
- Crisis generalizadas: pérdida total y absoluta de la conciencia o de la conexión del medio desde el inicio de la crisis.
La segunda característica que define a una crisis epileptica es la semiologi?a cli?nica durante el episodio (Blume et al., 2001), pudiendo asociarse en una misma crisis más de una manifestación. Las principales son:
- Motoras: contracciones musculares repetidas (que si son amplias y en salvas se llaman clonías, y si son muy breves, mioclonías); con hipertonía (y se denominan entonces crisis tónicas), o con pérdida del tono muscular y caída brusca al suelo (crisis atónicas).
- Sensitivas: alteraciones de la percepción en cualquiera de los cinco sentidos o síntomas autonómicos.
- Psíquicas: miedo, depresión, alegría, alucinaciones, ilusiones o experiencias de tipo déjà-vu, jamais-vu.
- Automatismos: actividad motora involuntaria consistente en movimientos repetitivos y estereotipados sin objeto (masticación, deglución, golpeteo, abrochado de botones, emisión de sonido, etc.).
- Aura: sensación subjetiva que experimenta el paciente antes de una crisis.
«En relación con la semiología clínica durante las crisis epilépticas, los autores nos refieren las principales manifestaciones clínicas que pueden aparecen en una crisis. Hay que tener en cuenta que estas manifestaciones pueden darse más de una a la vez, en la crisis epiléptica. Para las manifestaciones clínicas psíquicas tenemos la aparición de alucinaciones, miedos, alegría… y también los fenómenos de déjà vu/jamais-vu que se refieren a situaciones en los que la persona cree haber vivido ya una situación que vive en presente y lo contrario, no haber vivido o no conocer situaciones o personas que sí debería reconocer». Dra. del Cerro
Por lo tanto, las crisis se definen y clasifican aunando ambas categorías clínicas:
- Crisis parciales simples o complejas (con o sin generalización secundaria), de semiología motora o sensitiva o psíquica, con o sin aura y con o sin automatismos.
- Crisis generalizadas (exclusivamente de semiología motora): crisis tónico-clónica generalizada, tónica o mioclónica. Un tipo peculiar de crisis generalizada es la ausencia, en la que no sucede ningún fenómeno, ni motor ni de alteración del tono, y el paciente permanece “desconectado” del medio sin más síntomas, para inmediatamente después, al ceder la crisis, continuar con la actividad que estaba realizando, sin el período poscrítico habitual (somnolencia, confusión) propio de los otros tipos de crisis.
SÍNDROMES EPILÉPTICOS
En la definicio?n de un si?ndrome epile?ptico, el tipo de crisis epile?ptica constituye un factor ma?s, de manera que para caractenzarlo se necesitan tambie?n datos complementarios de la evolucio?n del cuadro y de otras manifestaciones cli?nicas. Enmarcar a un paciente en un determinado sindrome epile?ptico aporta mayor informacio?n en cuanto al prono?stico y al tratamiento. El principal factor que caracteriza el diagnóstico de un síndrome epiléptico en neuropediatría es la edad del niño. Asociados a las diferentes etapas de maduración cerebral, se observan síndromes específicos y característicos de cada edad que sólo se dan en esos períodos de la vida y no en otros.
Síndromes epilépticos del período neonatal
Las convulsiones en el recién nacido difieren de las del niño mayor en varios aspectos, principalmente debido a la dificultad para catalogar los fenómenos paroxísticos neonatales en relación con la expresividad clínica limitada del cerebro inmaduro, siendo escasas las correlaciones clínica-EEG (Roldán, 2005).
Síndromes epilépticos del lactante y el niño pequeño
Síndrome de West
Descrito por West en su propio hijo de 4 meses. Se conforma por la siguiente tríada: espasmos infantiles, hipsarritmia y deterioro psicomotor.
El síndrome de West tiende a presentarse antes de los 12 meses de edad, con un máximo en torno a los 6 meses. Son raros los casos que se inician después de los 24 meses. Una característica peculiar es que, cuando aparecen las crisis epilépticas, con frecuencia hay una detención o regresión en el desarrollo psicomotor, el niño puede perder la sonrisa social, el interés por el entorno e incluso hitos evolutivos ya alcanzados. Las Crisis epilépticas típicas son los espasmos infantiles, que consisten en contracciones tónicas (sostenidas) de la cabeza, el tronco y las extremidades, bilaterales, en general simétricas, bruscas, de aproximadamente 1 segundo de duración. Pueden producir una flexión o extensión del tronco y las extremidades, o una combinación de ambos (recuerdan un movimiento de «abrazo»). Ocurren en salvas de hasta más de 100 elementos, varias veces al día (predominantemente al despertar o al conciliar el sueño).
En el electroencefalograma (EEG), el patrón característico es la hipsarritmia. Ésta se define, en vigilia, por una mezcla anárquica y aleatoria de ondas lentas de alto voltaje y puntas de amplitud y localización cambiante en todas las áreas corticales, sin sincronía entre ambos hemisferios ni ritmo basal discernible.
Síndrome de Lennox-Gastaut
El Síndrome de Lennox-Gastaut se caracteriza por la tríada:
- Ausencias atípicas, crisis tónicas y crisis atónicas.
- Patrón en el EEG con descarga de punta-onda lenta generalizada.
- Discapacidad intelectual.
El 20% de los casos previamente presenta un síndrome de West. La edad de inicio se sitúa entre los 2 y los 5 años (con un máximo entre los 3 y los 5 años). Las crisis pueden ser de varios tipos (las tónicas son las más frecuentes, hasta un 92%). Aunque aparecen en vigilia, lo que caracteriza al síndrome es que suceden durante el sueño. Cursan con afectación neuropsicológica (cognitiva y conductual) y se caracterizan por su refractariedad y por su etiología (aunque algunos casos se consideran idiopáticos o criptogénicos, al no hallarse una causa concreta).
«El Síndrome de Lennox-Gastaut presenta crisis ya en niños muy pequeños (16 meses de edad, aunque el texto indica los 24 meses como inicio) y la tonicidad suelen darse con más frecuencia cuanto más jóvenes son los pacientes. Las caídas en las crisis tónico-clónicas se dan más tarde (4-5 años de edad media). La afirmación que Vd. señala como contradictoria no lo es: … «Aunque aparecen en la vigilia, lo que caracteriza al síndrome de Lennox-Gastaut es que suceden durante el sueño» , los autores están describiendo algo muy frecuente en estos niños y es que, a pesar de que en la mayoría de ellos aparecen durante la vigilia la inmensa mayoría de los casos (95%)tienen varios tipos de crisis y en más de la mitad de ellos tienen crisis tónicas nocturnas por lo que con frecuencia pueden pasar desapercibidas. Durante el día, la crisis tónicas (pérdida repentina de consciencia con contracción muscular generalizada de todo el cuerpo e hiperextensión brusca) obliga a que estos niños lleven permanentemente un casco para protegerlos de la multitud de golpes que pueden sufrir». Dra. del Cerro
Síndromes epilépticos del niño mayor y el adolescente (Ruiz-Falcó et al., 2008)
Epilepsia con ausencia
Antiguamente denominada «petit mal«,cursa con ausencias y comienza entre los 3 y los 12 años de vida. Si se inicia entre los 3 y los 9 años hablamos de Epilepsia con ausencias infantiles y si aparece a partir de los 9-10 años, Epilepsia con ausencias juveniles. En el 40% de los casos se asocian a crisis tónico-clónicas generalizadas, usualmente entre los 10 y los 15 años de edad. Se observa un EEG característico: paroxismos generalizados de punta-onda a 3 Hz. Las crisis se desencadenan de forma característica con la hiperventilación. La respuesta al tratamiento es muy buena, aunque de forma infrecuente aparecen casos refractarios.
Epilepsia parcial benigna con paroxismos rolándicos o centrotemporales
Representa el síndrome epiléptico benigno infantil por excelencia. Las crisis se inician entre los 3 y los 13 años. Aunque pueden aparecer diferentes tipos de crisis parciales con o sin generalización secundaria, la crisis típica se da durante el sueño (primeras o últimas fases de éste) y es una crisis parcial simple con semiología hemifacial: clonías, desviación de la boca y parestesias, manifestaciones orofaringolaríngeas y anartria. Por lo general son muy breves (menos de 2 minutos), y en la mayoría de las ocasiones muy poco frecuentes o de crisis única.
El diagnóstico lo ofrece la localización de la descarga EEG en el área centrotemporal o rolándica. El pronóstico suele se excelente y, dada la benignidad y la escasa recurrencia, no está indicado instaurar tratamiento en el inicio.
Sindromes epile?pticos con punta-onda continua durante el suen?o (POCS)
Los POCS están definidos por el EEG en el sueño de punta-onda generalizada a 1,5-2 Hz de forma continuada.
El criterio diagnóstico clásico es que la punta-onda ocupe al menos el 80% del total del sueño no REM (en discusión). En vigilia, los enfermos pueden presentar un EEG normal o alteraciones focales, a veces iguales que en la epilepsia parcial benigna con paroxismos rolándicos. Aproximadamente el 40% manifiesta anomalías neurológicas, tratándose entonces de sintomáticas, y el resto muestran normalidad neurológica antes de la aparición de la POCS. Se presentan con crisis de cualquier semiología (excepto to?nicas) o sin crisis. Otra caracteristica del POCS son los síntomas neuropsicológicos (trastornos de conducta, dificultades de aprendizaje y de lenguaje, síntomas compatibles con TEA, TDA con o sin hiperactividad, etc.). Las crisis suelen ser refractarias al tratamiento. Es preciso que remitan las POCS, para lo que se precisa la administración de corticosteroides durante varios meses. En un período variable entre 2 y 7 años del inicio, se logra el control de las crisis y de las POCS, aunque las secuelas neuropsicológicas pueden ser ya permanentes.
Existen tres síndromes de POCS: Síndrome de Tassinari, Epilepsia afasia adquirida (síndrome de Landau-Kleffner) y la Epilepsia parcial benigna atípica de Aicardi.
COMORBILIDAD CON OTROS TRASTORNOS
En los niños con epilepsia se aprecia una mayor incidencia de discapacidad intelectual, trastorno del lenguaje, dificultades de aprendizaje y problemas de integración social. Todavía hay controversia en cuanto a si las crisis epilépticas en sí mismas pueden producir el grado de deterioro mental que se observa, o si debe tenerse en cuenta la etiología, la medicación, los factores psicosociales y el tipo de síndrome epiléptico. Existe un leve aumento de la mortalidad en relación con la enfermedad subyacente, y riesgo de ahogamiento y de muerte súbita.
Son muchos los trastornos orgánicos de la edad pediátrica en los que la epilepsia es uno de los principales síntomas y que supone uno de los condicionantes más relevantes de la gravedad y del pronóstico del proceso (Parra e Iriarte, 2007).
VALORACIÓN DIAGNÓSTICA
Se basa en una minuciosa historia clínica, que constituye la base del diagnóstico y será la que aporte las claves no sólo para diagnosticar la crisis, sino para encuadrar el tipo de síndrome epiléptico y plantear el esquema terapéutico, así como el diagnóstico diferencial apropiado (Arrabal et al., 2013)
Anamnesis
El diagnóstico de epilepsia se basa fundamentalmente en la historia clínica, en la que cobran especial importancia las descripciones de los fenómenos que suceden durante la crisis de la manera más detallada y precisa posible, así como la evolución: si hay afectación de la conciencia desde el inicio hasta el final, la secuencia de aparición de la semiología clínica y su progresión (tono muscular, fenómenos motores y sensitivos, automatismos, etc.), la presencia de aura, mordedura de lengua o pérdida del control de esfínteres, así como la finalización del episodio y la recuperación poscrítica.
Es importante recoger también información acerca de antecedentes personales (parto, desarrollo psicomotor, enfermedades sistémicas, etc.) y familiares (enfermedades neurológicas, epilepsia, etc.), que proporcionan datos fundamentales acerca de posibles etiologías.
Exploración física
Debe ser general y completa. Debe hacer constar la presencia de rasgos dismórficos o discromías. La exploración neurológica será asimismo sistematizada, completa y minuciosa.
«La posible importancia de la presencia de rasgos dismórficos (anomalías craneofaciales)- o discromías (alteraciones en tono de la piel) son importantes hacerlas patentes en la exploración de niños con posible valoración diagnóstica de EPILEPSIA infantil debido a los múltiples síndromes que cursan con estas alteraciones craneofaciales, y epidérmicas, que pueden coadyuvar un trastorno neurológico relacionado o no? con la posibilidad del Diagnóstico de Epilepsia infantil. Le sugiero una revisión muy específica sobre este tema de J.M. Santolaya. «Rasgos dismórficos que implican alteración neurológica. Pautas de Actuación».REV NEUROL , 35:58-67, 2002. DOI: https://doi.org/10.33588/rn.3501.2002149″. Dra. del Cerro
Pruebas complementarias
En general no son necesarios estudios analiticos; se solicitan cuando se trata de investigar procesos específicos relacionados con la etiología de las crisis o de epilepsias sintomáticas (secundarias a una afectación principal como cromosomopatías, enfermedades metabólicas, etc.).
El EEG debe realizarse en todo paciente con una crisis no provocada, aunque hay que recordar que el diagno?stico de crisis epile?ptica es clínico y un EEG normal no lo descarta. De la misma forma, un EEG patológico no supone necesariamente un diagnóstico de epilepsia: el 10-15% de la población presenta alteraciones inespecíficas en el registro, y el 1% muestra anomalías epileptiformes sin crisis. Si el EEG de vigilia es normal pero se mantiene la sospecha de crisis epilépticas que pueda conformar un síndrome epiléptico, debe realizarse un EEG en privación de sueño y durante el sueño, ya que estas dos circunstancias son las principales activadoras de descargas epilépticas. Puede ser precisa una monitorización vídeo-EEG de más de 24 horas (imprescindible en la evaluación prequirúrgica de la epilepsia). El electroencefalograma (EEG) es la principal prueba para la clasificación de los síndromes epilépticos, así como para establecer el diagnóstico diferencial.
Entre las pruebas de neuroimagen, la resonancia magnética funcional (RMf) se utiliza principalmente en el ámbito de la cirugía de la epilepsia (Koepp y Woermann, 2005). La tomografía por emisión de positrones PET y la tomografía Computarizada por Emisión de Fotón Único SPECT no son pruebas rutinarias, aunque se precisan ante determinadas epilepsias refractarias y en estudios prequirúrgicos de la epilepsia.
INTERVENCIÓN
Tratamiento agudo
Ante una crisis epiléptica se recomienda poner al paciente en posición de seguridad para evitar la aspiración y posibles lesiones. No hay que forzar la apertura de la vía aérea (boca) ni introducir objetos en ella (dedos, palos, pañuelos, etc.), ya que el riesgo de aspiración y lesión es mayor que cualquier posible y más que dudoso beneficio. Si la crisis es generalizada y no cede espontáneamente, se puede administrar una canuleta de diazepam rectal o una instilación de midazolam en la mucosa bucal.
Tratamiento crónico con fármacos antiepilépticos
El tratamiento debe ser perfectamente individualizado y consensuado con la familia, valorando con cuidado sus riesgos y beneficios (Panayiotopulos, 2007). Los principales factores que deben ser considerados a la hora de instaurar un tratamiento con antiepilépticos son:
- Seguridad del diagnóstico.
- Tipo de síndrome epiléptico.
- Riesgo de recurrencia de las crisis y sus consecuencias.
- Eficacia del fármaco y efectos secundarios.
- Opinión de los padres.
Como norma general, no suele iniciarse un tratamiento en una primera crisis epiléptica idiopática o probablemente sintomática hasta el segundo episodio y, en determinados síndromes (ej. epilepsia benigna de la infancia), se recomienda como norma no tratar.
El objetivo del tratamiento es conseguir el control absoluto de las crisis sin provocar efectos secundarios. En algunos síndromes epilépticos catastróficos o especialmente refractarios este objetivo no es una expectativa real. En una primera aproximación, siempre se intenta la monoterapia, aunque con relativa frecuencia es necesario recurrir a distintas combinaciones de fármacos, en especial en aquellos síndromes refractarios en los que concurren diferentes tipos de crisis (Rolda?n et al., 2009).
Otros tratamiento no farmacológicos
Dieta cetogénica: 80% de grasas, 15% de proteínas y 5% de hidratos de carbono. Induce la formación de cuerpos cetónicos que son semejantes estructuralmente al ácido ?-aminobutírico (GABA), uno de los principales neurotransmisores implicados en el control de la epileptogénesis. Es especialmente útil y hasta necesaria en algunos errores congénitos del metabolismo.
Estimulador del nervio vago: actúa sobre circuitos necesarios para la sincronización de las crisis. Está indicado en epilepsias refractarias no candidatas a cirugía.
Bisturí gamma: administración focal de radiación ionizante (radiocirugía). Indicado en hamartomas hipotalámicos o malformaciones vasculares.
Cirugía de la epilepsia: sólo en algunos casos de epilepsias refractarias a otros tratamientos.
Hábitos de vida
Los niños epilépticos deben llevar una vida normal para su edad, lo que incluiría evitar la ingesta de alcohol y la privación de sueño. En aquellos síndromes que cursan con fotosensibilidad, habrá que ser cautos con los videojuegos. Pueden practicar cualquier tipo de deporte, aunque en la natación se recomienda supervisión.
REPERCUSIONES SOCIALES
La experiencia psicológica y social de la epilepsia supone menor independencia, restricciones en actividades frecuentes, hospitalizaciones, estigma y aislamiento social; es decir, es un síndrome con el potencial de limitar el desarrollo y afectar de manera negativa la calidad de vida. Cuando el ambiente familiar está marcado por excesivo temor, preocupación, vergüenza o inseguridad, el niño integra en su vida un sentimiento de incapacidad. Todos estos desajustes se multiplican y dramatizan en el caso de epilepsias de difícil manejo, y más todavía cuando el niño tiene una discapacidad añadida. Por ello, es necesario brindarles (tanto a las familias como a los niños) herramientas para enfrentar, aceptar y participar en el control del problema.
La escuela es el elemento fundamental para la formación, el aprendizaje, la educación y la integración social. El niño debe seguir la misma escolarización que le corresponda a un niño no epiléptico acorde a su capacidad. Los niños epilépticos que además tienen trastornos neuropsicológicos (déficits cognitivos y emocionales, de conducta, sociabilidad, etc.) necesitarán una adaptación curricular y modelos pedagógicos multidisciplinares.
Adema?s del abordaje de la epilepsia dentro de la familia, e?sta tambie?n se enfrenta al estigma social que acompai?ia a la enfermedad. Según datos de la ILAE y la OMS, en algunos países de África y Asia se sigue identificando a los pacientes epilépticos como poseídos por espíritus y hechizados. En China y EE.UU., en la década de los 90, un 20% de los entrevistados no daría empleo a estos pacientes y no querrían estar emparentados con ellos. En EE.UU., hasta 1980 se les prohibía el matrimonio y hasta 1970 no se legalizó la entrada de las personas epilépticas a restaurantes, teatros y centros de ocio. En India y China hoy día la epilepsia es causa de anulación de matrimonio.
RESUMEN
La epilepsia en la infancia constituye una entidad heterogénea y compleja. Engloba diferentes síndromes determinados por la edad del niño, la tipología crítica y el registro EEG que los caracteriza. El tipo de síndrome epiléptico y su etiología marcarán la evolución natural de la enfermedad.
El diagnóstico se basa fundamentalmente en la historia clínica. La principal prueba complementaria es el registro EEG; pueden ser necesarias otras de segundo escalón, como las pruebas de neuroimagen (estructural o funcional) y estudios genéticos o metabólicos en función de la etiología y la evolución.
El tratamiento farmacológico debe ser perfectamente individualizado y consensuado con la familia, valorando con cuidado sus riesgos y beneficios. En la actualidad se dispone de numerosos fármacos antiepilépticos que se deben seleccionar y combinar en función del síndrome epiléptico y las características individuales de cada niño y familia. Se dispone, además, de otras alternativas terapéuticas no farmacológicas y de opciones quirúrgicas.
La epilepsia infantil repercute inevitablemente sobre la vida social y académica del niño, así como sobre la dinámica familiar. Dicha repercusión estará determinada por el tipo de síndrome epiléptico y por el manejo que del proceso hagan los diferentes profesionales implicados. El adecuado conocimiento de la epilepsia evitará impactos negativos carentes de fundamento, heredados de conceptos erróneos y estigmatizantes hacia estas personas.
ESQUEMA
AUTOEVALUACIÓN
En exámenes anteriores preguntaron…
| La clasificación más empleada, en la práctica habitual, de las crisis epilépticas en relación a la afectación de la conciencia en su transcurso, diferencia dichas crisis en Focales (antes parciales) y Generalizadas. |
| La prevalencia general de la epilepsia infantil es del 0,4-0,8%, lo que supone una incidencia de 20-70/100.000/año. |
| Síndrome epiléptico: Grupo de signos y síntomas que configuran un trastorno epiléptico único. |
| Epilepsia: Trastorno cerebral caracterizado por una predisposición duradera a generar crisis epilépticas y por las consecuencias neurobiológicas, cognitivas, psicológicas y sociales de esta condición. |
| Crisis Epiléptica. Ocurrencia transitoria de signos y/o síntomas derivados de una actividad anormal excesiva o síncrona de la actividad neuronal. |
| La principal prueba complementaria en el diagnóstico de la Epilepsia infantil, además de la historia clínica es el EEG. |
| La valoración diagnóstica de la Epilepsia se basa, fundamentalmente en una minuciosa historia clínica. |
| Entre los tratamientos no farmacológicos de la epilepsia podemos citar el estimulador del nervio vago. |
| Puede remitir espontáneamente en el 20-30 % de los casos. |
| Síndrome epiléptico benigno infantil por excelencia: Epilepsia parcial benigna con paroxismos rolándicos o centrotemporales. |
REFERENCIAS
- Arnedo Montoro, M. (2018). Neuropsicología del desarrollo. Madrid: Médica Panamericana.
- Apuntes CARMEN ORTEGO
- YouTube