

Versión 2
Dimensiones sintomáticas en la esquizofrenia
Síntomas positivos y negativos de la esquizofrenia
El síndrome de la esquizofrenia consiste en una mezcla de síntomas que normalmente se dividen en dos categorías mayores: positivos y negativos. Síntomas positivos, como los delirios y las alucinaciones, que reflejan el desarrollo de los síntomas de psicosis y que pueden llegar a ser tan dramáticos como para dar lugar a una pérdida de contacto con la realidad. Los síntomas negativos reflejan una pérdida de funciones y sentimientos normales, como la pérdida de interés en las cosas o la incapacidad de síntomas experimentar placer.
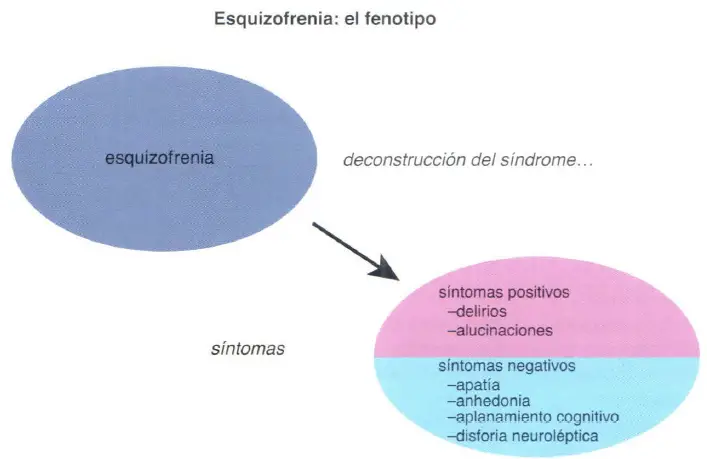
Algunos síntomas negativos de la esquizofrenia, tales como un discurso reducido, aspecto descuidado y la evitación del contacto ocular pueden identificarse exclusivamente mediante la observación del paciente.

Otros síntomas negativos de la esquizofrenia pueden identificarse mediante simples pregustas. Por ejemplo, un cuestionario breve puede revelar el grado de respuesta afectiva, el nivel de interés en aficiones o en la consecución de metas, y el deseo de iniciar y mantener contactos sociales.

¿Es la esquizofrenia una enfermedad neurodegenerativa?
En la esquizofrenia, no hay acuerdo absoluto en asuntos de neurociencia, pues no es ciencia matemática. Está claro que parece haber una disposición genética, con malformaciones o conexiones aberrantes en el desarrollo neuroevolutivo, y que suelen ser los cambios hormonales durante la pubertad y más allá los detonantes de la activación de la enfermedad.
Otra cosa es que el trastorno como tal no conlleve neurodegeneración asociada (algunos estudios con resonancia magnética nuclear sí lo demuestran; otros, no), especialmente en los casos en los que no se aplica tratamiento farmacológico; sobre todo, si se tiene en consideración las últimas investigaciones que hipotetizan una participación muy activa de hiperexcitabilidad neuronal (hecho que sí se sabe que conduce a la destrucción celular) inducida por la hiperactivación del sistema glutamatérgico.
Y, sin afirmar ni negar al respecto, pues los expertos no se ponen del todo en acuerdo, se aportan algunos resúmenes de revisiones teóricas en revistas especializadas de impacto científico relevante (de la misma fecha, 1998; y otras más recientes aún) que no consideran la posibilidad de que la esquizofrenia sea una enfermedad degenerativa en sentido estricto (en todo caso, sin tratamiento, sí se empeora con el tiempo, ¿¿degenera??, aunque tampoco es así en todos los casos), pero tampoco un «error» neuroevolutivo de forma aislada; sino que podría ser -como afirman Gross y Huber (2008)- procesos cerebrales regresivos (involutivos) especiales.
[Consultar más aquí]
[Consultar más aquí]
[Consultar más aquí] (Gross y Huber, 2008).
No entiendo bien la hipótesis de la hipofunción de los receptores NMDA en la esquizofrenia
Esta hipótesis indica que la sintomatología de la esquizofrenia es debida a una hipofunción de los receptores glutamatérgicos NMDA en el cerebro. Esta hipofunción sería la responsable de la aparición de los síntomas positivos y negativos característicos de la esquizofrenia.
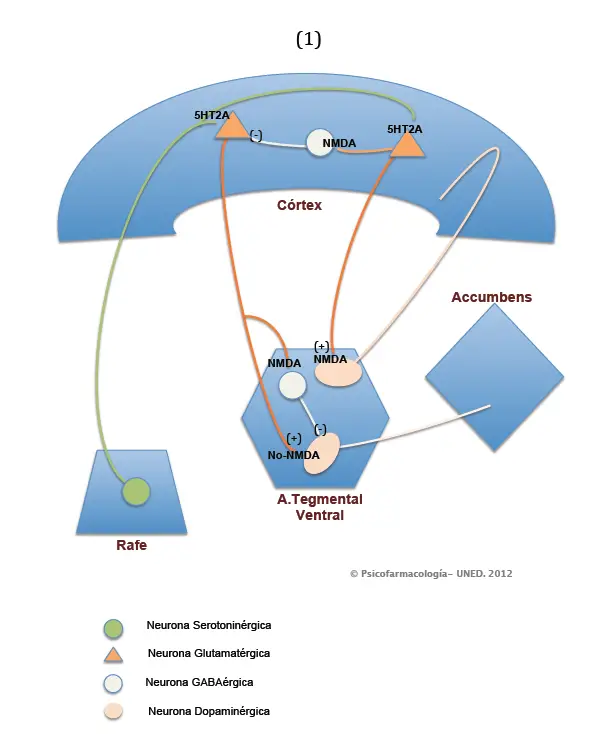
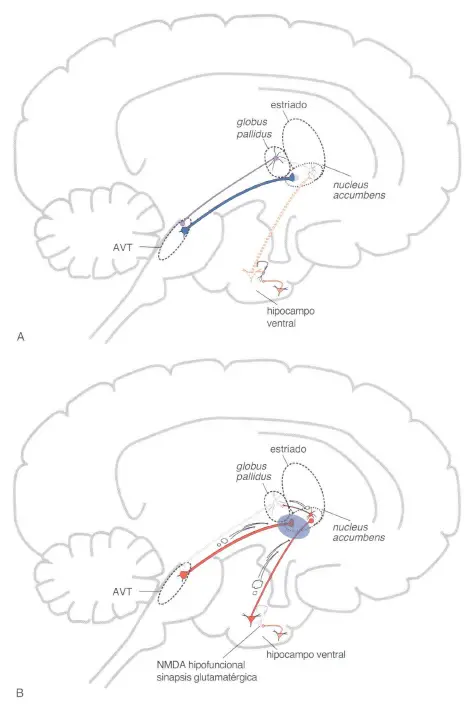
Tal y como se muestra en el Gráfico 1, que se incluye seguidamente, la actividad de las neuronas piramidales de la capa media de la corteza -que son glutamatérgicas- estaría regulada por la actividad de interneuronas GABAérgicas, que poseen receptores glutamatérgicos NMDA, gracias a lo cual pueden recibir inputs de estas neuronas piramidales (parte superior del gráfico). Es decir, la actividad de estas neuronas piramidales estaría «autorregulada» mediante interneuronas GABAérgicas, que actuarían, a modo de freno, sobre la propia actividad de ellas, de las neuronas glutamatérgicas.
A su vez, estas neuronas piramidales glutamatérgicas, proyectarían hacia el área tegmental ventral (ATV). Ahí harían sinapsis directa con neuronas dopaminérgicas y con otras neuronas GABAérgicas. Algunas de estas neuronas dopaminérgicas poseerían receptores NMDA, mientras que otras no (serían receptores glutamatérgicos No-NMDA, como, por ejemplo, Kainato o AMPA).
Las neuronas dopaminérgicas con receptores No-NMDA, proyectaría hacia la zona ventral del núcleo estriado, en concreto, sobre el núcleo accumbens (vía mesolímbica). Por su parte, las neuronas dopaminérgicas con receptores NMDA lo harían hacia el córtex prefrontal, constituyendo parte de la vía mesocortical.
Al igual que la actividad de las neuronas glutamatérgicas de la corteza está regulada por interneuronas GABAérgicas que actuarían como un sistema de freno sobre ellas, la actividad glutamatérgica de estas neuronas piramidales dentro del ATV, también estaría regulada, mediante interneuronas GABAérgicas presentes en esta estructura (ver la parte inferior del gráfico: A. Tegmental Ventral). Así, la actividad de las neuronas dopaminérgicas del ATV estaría regulada de dos formas. Una, indirecta mediante el freno que las neuronas GABAérgicas ejercerían sobre las piramidales glutamatérgicas en la corteza y, dos, directa por la actividad GABAérgica dentro del ATV, neuronas éstas que recibirían, a su vez, input de la neuronas piramidales de la corteza.
De estos dos tipos de regulación dopaminérgica (aunque Stahl da predominio al control en el ATV por parte de las neuronas GABAérgicas), recientes estudios indican que puede resultar más importante la regulación GABAérgica que las interneuronas GABAérgicas ejercen sobre las neuronas piramidales en el córtex cerebral, que aquél que se realiza dentro del ATV ( J. T. Coyle y cols.. Beyond the dopamine receptor: novel therapeutic targets for treating schizophrenia. Dialogues in Clinical Neuroscience. Vol. 12 (3) . 2010. 359-382).
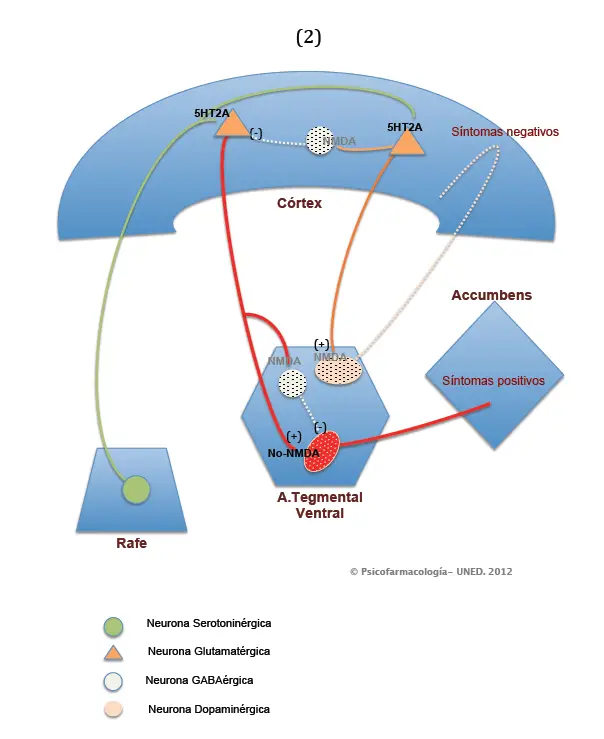
La hipótesis de la hipofunción de receptores NMDA en la esquizofrenia supondría una desregulación o una hipofunción de este tipo de receptor en aquellas células en las que estuviera presente. Así, afectaría al input que recibirían las neuronas GABAérgicas por parte de las glutamatérgicas, tanto en el córtex como en el ATV, y al de las neuronas dopaminérgicas del ATV con receptores NMDA, que, recibiendo información directa de las neuronas piramidales de la corteza, proyectarían hacia el córtex prefrontal, constituyendo, como ya se ha dicho, parte de la vía mesocortical.
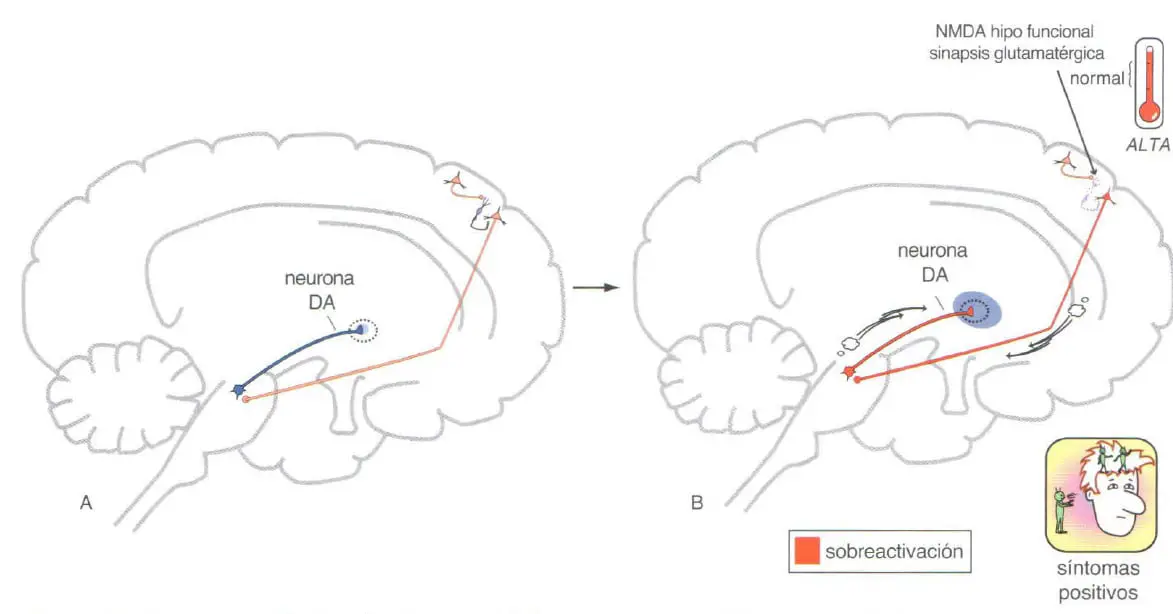
Siendo esto así, en el cerebro de una persona afectada de esquizofrenia, las neuronas GABAérgicas del córtex no podrían ejercer su acción de «freno» sobre las neuronas piramidales glutamatérgicas en el córtex, ni sobre las dopaminérgicas en el ATV. El resultado de todo ello sería una sobreactivación de las neuronas glutamatérgicas de la corteza, que conllevaría una sobreexcitación de las neuronas dopaminérgicas del ATV que proyectan sobre el núcleo accumbens (ver Gráfico 2).
Esta sobreexcitación de las neuronas dopaminérgicas, se piensa que es la causante de la aparición de la sintomatología positiva en la esquizofrenia.
Por su parte, dado que las neuronas dopaminérgicas del ATV que poseen receptores NMDA tampoco podrían responder adecuadamente, el output que enviarían al córtex prefrontal sería deficiente, lo que daría lugar a la aparición de los síntomas negativos.
Nótese en la siguiente figura (2), que las líneas marcadas en rojo indican sobreexcitación, mientras que las discontinuas muestran que estas neuronas están desactivadas o su activación es deficiente.
¿Son los síntomas negativos de la esquizofrenia la consecuencia de la deficiencia funcional de la vía mesolímbica o, por el contrario, el resultado de la hiperactivación de dicha vía?
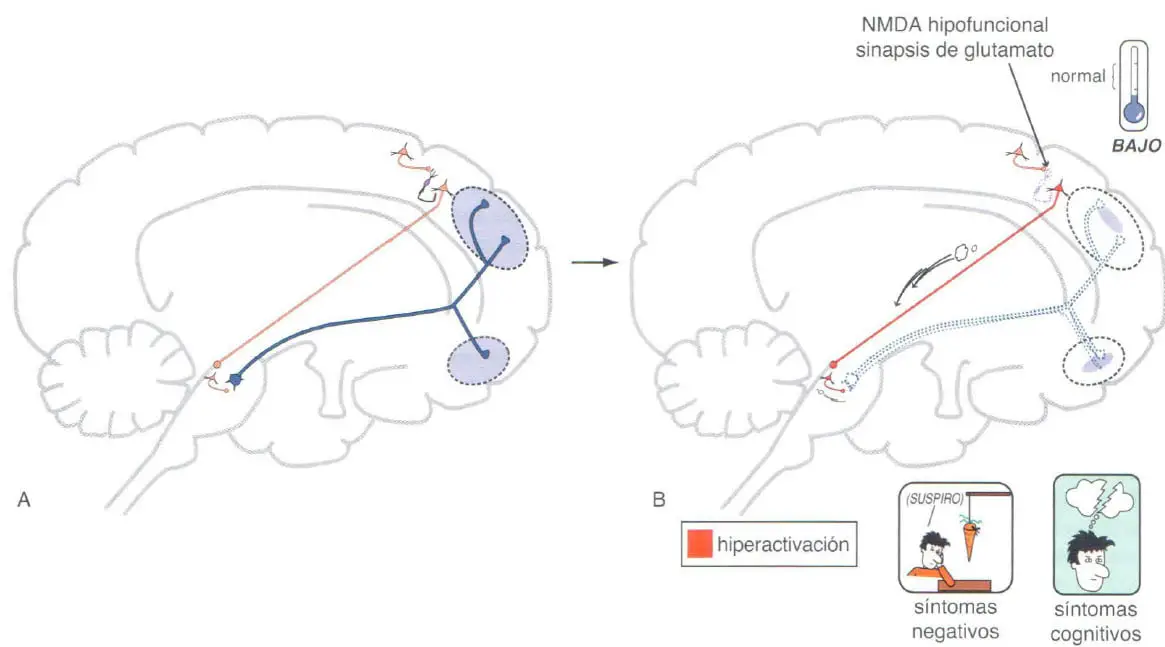
Según la hipótesis de hipofunción de la vía dopaminérgica mesocortical (que proyecta sus terminales al córtex prefrontal dorsolateral -relacionado con aspectos cognitivos- y al córtex prefrontal ventromedial -relacionado con aspectos afectivos- conlleva el consiguiente aumento de los síntomas negativos en los que están involucradas las dos ramas de proyección cortical de la vía mesocortical.
No obstante, en algunos pacientes esquizofrénicos, se ha postulado también que los síntomas negativos (desmotivación exacerbada, falta de interés en relaciones sociales, falta de higiene, etc.), podrían estar relacionados no sólo con déficit de la vía dopaminérgica mesocortical sino también con la deficiencia -y no hiperactivación- funcional de la vía mesolímbica, en tanto en cuanto a su implicación con el sistema de recompensa cerebral. Ello puede parecer contradictorio con lo expuesto en el texto previamente; pero, por otra parte, muchos especialistas consideran desde un punto de vista neurobiológico el circuito mesocorticolímbico como algo global, es decir, que no son consideradas vías aisladas, y que el trastorno ha de considerarse como una desregulación del sistema dopaminérgico cerebral no focalmente sino completo, precisamente por sus interacciones con otros sistemas de neurotransmisión en diferentes regiones cerebrales en donde se establecen las interconexiones neurales que dan lugar a los diversos aspectos de los síntomas positivos y negativos.
Asimismo, algunos autores defienden que algunas neuronas de la vía mesolímbica -por conexiones aberrantes, propias del trastorno, durante el desarrollo- se pueden hiperactivar mientras que otras selectivamente se vuelven hipofuncionales, dependiendo de las aferencias diversas que les llegan de otros sistemas de neurotransmisión (serotoninérgico, GABAérgico, glutamatérgico, opioide, etc.). Es bastante probable que las alteraciones cognitivas y afectivas, y de recompensa, en la esquizofrenia sean el resultado de anomalías globales del sistema corticomesolímbico derivadas de las complejas interacciones entre los diferentes sistemas de transmisión señalados. De hecho, cuando se estudia los mecanismos neurobiológicos de recompensa de las drogas (capítulo 14), se entiende mejor dicha complejidad, habida cuenta de las conexiones corticales con núcleos subcorticales -como el n. accumbens y la amígdala- desde la corteza prefrontal, la cual ejerce un control «reflexivo» del consumo de sustancias.
Como complemento a lo anterior, es habitual que en la esquizofrenia se presente abuso de drogas (tabaco, alcohol, estimulantes, etc.), lo que ha venido en denominarse patología dual (es decir, un trastorno psicótico más el componente de conductas adictivas a sustancias o de drogadicción). Esto está relacionado precisamente con la pregunta formulada, en el sentido de que la vía mesolímbica o de la recompensa está también -en mayor o menor medida- alterada en la esquizofrenia. Es por ello que los pacientes con esquizofrenia suelen tener sintomatología de desmotivación y anhedonia, y es precisamente esto lo que a muchos de ellos les pueda inducir al consumo de drogas (considerada por algunos autores como «automedicación») que les devuelvan algo de «refuerzo» vital cuando los núcleos del placer están deteriorados en su función normal (presumiblemente una hipofunción dopaminérgica mesolímbica), y especialmente en pacientes con tratamiento farmacológico antipsicótico con potencialidad para la magnificación de síntomas negativos.
Sin embargo, la disminución de dopamina causante de anhedonia y disforia también puede ser previa al consumo de sustancias, y ser -o representar- un rasgo constitucional de vulnerabilidad para la adicción, lo cual hay que tenerlo en consideración a la hora de la prescripción de determinados tratamientos antipsicóticos.
El tratamiento de pacientes esquizofrénicos con antipsicóticos puede empeorar los síntomas negativos de la enfermedad ¿No es entonces contraproducente dicho tratamiento?
Los síntomas negativos se pueden magnificar o aumentar en esquizofrénicos por el tratamiento con antipsicóticos convencionales (neurolépticos), precisamente porque el bloqueo de los receptores D2 con estos fármacos es bastante potente en todas las vías dopaminérgicas. Se eliminan los síntomas positivos pero suelen aumentar los negativos.
No ocurre así con otros tratamientos, a base de antipsicóticos atípicos, los cuales pueden regular de manera más selectiva el control de la liberación de dopamina a través del bloqueo, por ejemplo, de los receptores 5-HT2 de serotonina, siendo la función de ésta inhibir la liberación de dopamina; de este modo, los antagonistas 5-HT2 pueden promover la liberación de dopamina en la vía mesocortical hipoactiva.
¿Sería correcto decir que los síntomas de agresividad de la esquizofrenia, además de ubicarse en el córtex orbitofrontal y la amígdala, también son provocados por el deficitario control serotoninérgico de la dopamina en la vía dopaminérgica mesolímbica?
Es más correcto afirmar -mejor que la ubicación en- «que en los síntomas agresivos propios de la esquizofrenia están involucrados o implicados el córtex orbitofrontal y la amígdala» (como en otros trastornos que cursan también con comportamiento agresivo por ausencia de control de impulsos fundamentalmente).
Por otra parte, es correcto afirmar que la agresividad en la esquizofrenia se produce o podría producir como resultado de un deficitario control serotoninérgico de la liberación de dopamina en la vía mesolímbica; es decir, por un aumento de liberación de dopamina en estructuras mesolímbicas, que están involucradas en la modulación de la conducta agresiva. De hecho, la lesión del área tegmental ventral (donde se encuentran localizadas los somas de las neuronas dopaminérgicas) y/o la disminución de serotonina reducen la agresividad en roedores; de otra parte, la estimulación de los receptores 5-HT2 produce la inhibición de liberación de dopamina al ser estimulada la función serotoninérgica, y promueve asimismo la eliminación de conductas agresivas tanto defensivas como de ataque.
Localización de los campos sintomáticos
Se cree que los diferentes campos sintomáticos de la esquizofrenia están regulados por regiones cerebrales únicas. Así, los síntomas positivos de la esquizofrenia están hipotéticamente modulados por una disfunción de circuitos mesolimbicos, mientras que los síntomas negativos están hipotéticamente originados por una disfunción de circuitos mesocorticales y también podrían implicar regiones mesolímbicas, como el núcleo accumbens, que es parte del circuito de recompensa del cerebro y juega un importante papel en la motivación. El nucleus accumbens también podría intervenir en el mayor indice de uso y abuso de sustancias observado en pacientes con esquizofrenia. Los síntomas afectivos están relacionados con el córtex prefrontal ventromedial, mientras que los síntomas agresivos (relacionados con el control de impulsos) se asocian con un procesamiento anormal de la información en el córtex orbitofrontal y la amígdala; los síntomas cognitivos se asocian con problemas en el procesamiento de la información en el córtex prefrontal dorsolateral. Aunque hay solapamiento de funciones entre las diferentes regiones cerebrales, la comprensión de qué regiones pueden desarrollar una función específica de forma predominante puede permitir un tratamiento más específico para cada perfil sintomático de cada paciente particular con esquizofrenia.
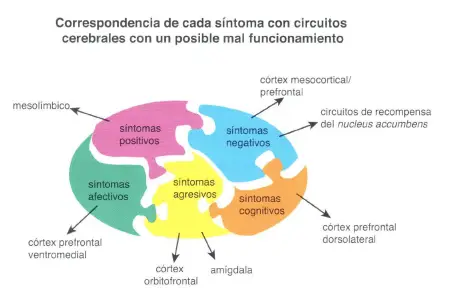
En la figura 4-4, página 84, se involucra a la zona mesolímbica en los síntomas positivos de la esquizofrenia y al córtex mesocortical/prefrontal y el núcleo accumbens para los síntomas negativos. No obstante, en la página 85 -segundo párrafo- explica que el núcleo accumbens está especialmente involucrado en los síntomas positivos. ¿Está el núcleo accumbens involucrado en ambos tipos de síntomas o es un errata?
No es una errata propiamente dicha. El texto en inglés está publicado en 2013 y nuestra edición traducida y actualizada es del 2016. Algo se sabía ya al respecto o al menos se intuía por trabajos especializados.
Hay que tener en cuenta que la neurociencia avanza de manera vertiginosa. Y más la psicofarmacología. Es por ello que en el texto, sin hacerlo de manera expresa, el autor introduce que el desequilibrio funcional del núcleo accumbens puede ser partícipe y responsable tanto de síntomas positivos como de los negativos. El cerebro es todo menos sencillo, y no hay zonas aisladas que exclusivamente participen o sean absolutas responsables funcionales de una conducta particular.
La estructura anatómica del núcleo accumbens indica que su función es la integración límbicomotora, por sus conexiones no sólo con el sistema límbico sino también con áreas corticales. Su función más relevante es en el circuito de recompensa. Sin embargo, a raíz de un estudio reciente, se ha descubierto que el accumbens se relaciona tanto con estímulos reforzantes como con los aversivos. En relación a este estudio, se descubrió que los niveles de dopamina en el accumbens pueden aumentar como respuesta a estímulos reforzantes y descienden drásticamente con los aversivos. De este modo, este núcleo en su comunicación con áreas corticales no sólo establece las bases para crear asociaciones entre las respuestas que nos proporcionan placer, sino que también influye en establecer otras asociaciones relativas a cómo evitar las que tienen consecuencias aversivas. Y dichas asociaciones se producen en la corteza.
Nunca hay que olvidar que las conexiones entre áreas son fundamentales en la integración y coordinación final del comportamiento, no sólo en lo que a respuestas emocionales se refiere sino a los aspectos cognitivos (estímulos negativos relacionados con la pregunta).
Neurotransmisores y circuitos en la esquizofrenia
La hipótesis principal de la esquizofrenia se basa en el neurotransmisor dopamina. Las neuronas dopaminérgicas utilizan la dopamina (DA) como neurotransmisor. Esta es sintetizada en los terminales nerviosos dopaminérgicos a partir del aminoácido tirosina, que es absorbido en la neurona desde el espacio extracelular y desde el torrente sanguíneo mediante una bomba de tirosina, o transportador.
Dopamina
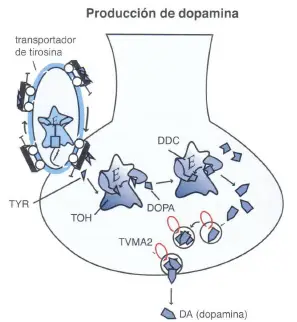
Síntesis de la dopamina. La tirosina (TYR), un precursor de la dopamina, es transportada al interior de los terminales nerviosos dopaminérgicos, vía transportador de tirosina, y convertida en DOPA, mediante la enzima tirosina hidroxilasa (TOH). La DOPA entonces es convertida en dopamina (DA) mediante la enzima DOPA decarboxilasa (DDC). Después de la síntesis, la dopamina es almacenada en vesículas sinápticas
gracias al transportador vesicular de monoaminas (TVMA2) y allí permanece hasta que es liberada en la sinapsis durante la neurotransmisión.
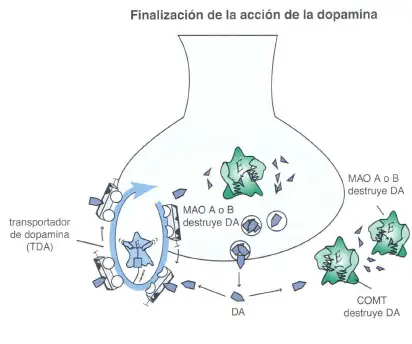
Finalización de la acción de la dopamina. La acción de la dopamina puede ser finalizada a través de múltiples mecanismos. La dopamina puede ser transportada fuera de la hendidura sináptica al interior de la presinapsis por medio del transportador de dopamina (TOA), donde puede ser realmacenada para un futuro uso. Alternativamente, la dopamina puede ser destruida en el espacio extracelular por la enzima catecol-0-metiltransferasa (COMT). Otras enzimas que pueden destruir la dopamina son la monoaminoxidasa A (MAO-A) y la monoaminoxidasa (MAO-B), que están presentes en la mitocondria dentro de la neurona presináptica y en otras células como las de la glía.
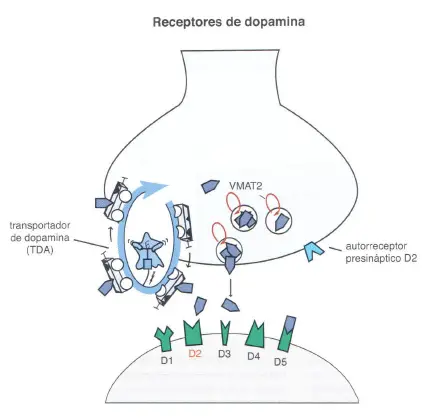
Receptores de dopamina. El transportador de dopamina (TOA) se encuentra en la presinapsis y es responsable de eliminar el exceso de dopamina en la sinapsis. El transportador vesicular de monoaminas (TVMA2) bombea dopamina al interior de las vesículas sinápticas para futuras neurotransmisiones. Hay, además, un receptor de dopamina del subtipo 2 en la presinapsis que funciona como autorreceptor, regulando la liberación de dopamina de la neurona presináptica. También hay varios tipos de receptores postsinápticos. Estos incluyen los receptores de dopamina de los subtipos 1, 2, 3, 4 y 5. Las funciones del receptor de dopamina-2 son las mejor estudiadas, porque es la zona principal de unión de prácticamente todos los agentes antipsicóticos así como de los agonistas dopaminérgicos usados en el tratamiento de la enfermedad de Parkinson.
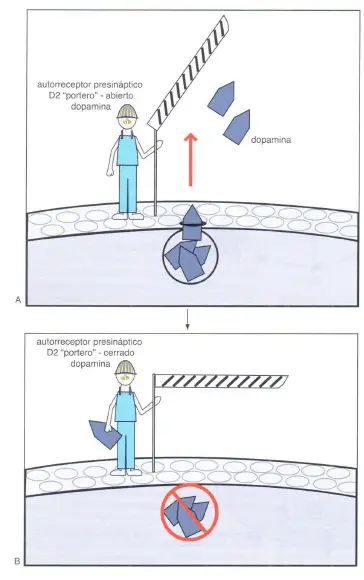
Autorreceptores presinápticos de dopamina 2 (D2). Los autorreceptores presinápticos D2 son como «porteros» de la dopamina. Es decir, cuando el receptor de la portería no está ligado a dopamina (no hay dopamina en la mano del portero), abren la barrera molecular, permitiendo la liberación de dopamina (A). Sin embargo, cuando la dopamina se une al receptor portero (ahora el portero tiene dopamina en la mano), cierran la barrera molecular e impiden la liberación de dopamina (B).

Autorreceptores presinápticos de dopamina-2. Los autorreceptores D2 pueden localizarse en el terminal axónico, como se muestra aquí. Cuando la dopamina se acumula en la sinapsis (A), se une al autorreceptor, que inhibe así la liberación de dopamina (B).
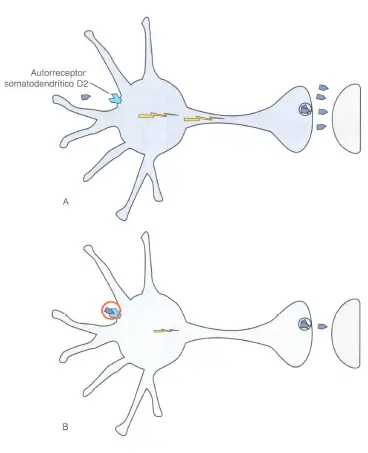
Autorreceptores somatodendríticos de dopamina-2. Los autorreceptores de dopamina-2 pueden también estar localizados en el área somatodendrítica, como se muestra aquí. Cuando la dopamina se une al receptor en esta área, se atenúa el flujo de impulsos en la neurona (véase la pérdida de impulsos eléctricos en la neurona en B), y esto detiene la liberación de más dopamina.
En ambos casos, la ocupación de los receptores D2 proporciona un fenómeno de retroalimentación negativo, o de freno, en la liberación de dopamina desde la neurona presináptica.
Principales vías dopaminérgicas del cerebro
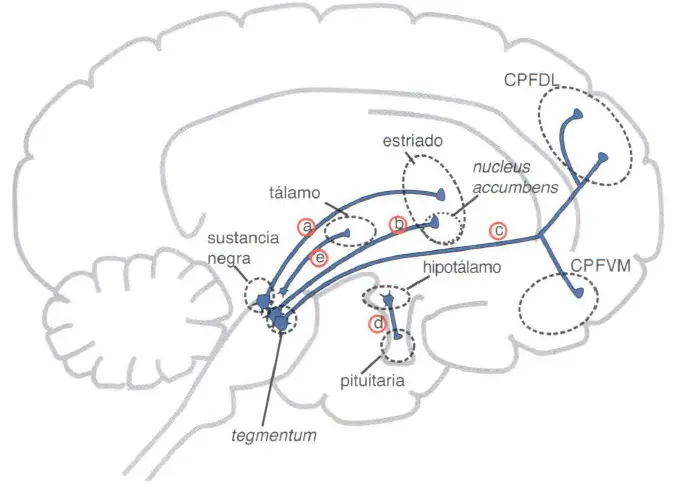
El conocimiento de la neuroanatomía de las vías neuronales dopaminérgicas en el cerebro puede ayudar a explicar los síntomas de la esquizofrenia, así como los efectos terapéuticos y los efectos secundarios de los fármacos antipsicóticos. Hay 5 vías dopaminérgicas principales:
- La vía dopaminérgica nigroestriada (a), que se proyecta desde la sustancia negra a los ganglios basales o estriado, es parte del sistema nervioso extrapiramidal y controla funciones motoras y movimiento.
- La vía dopaminérgica mesolímbica (b), se proyecta desde el área tegmental ventral del mesencéfalo al nucleus accumbens, una parte del sistema límbico del cerebro que se cree que gestiona múltiples funciones como las sensaciones placenteras, la potente euforia de las drogas de abuso así como la producción de delirios y alucinaciones en la psicosis.
- La vía dopaminérgica mesocortical (c), esta relacionada con la vía dopaminérgica mesolímbica. Se proyecta desde el área tegmental ventral del mesencéfalo a áreas del córtex prefrontal, donde puede desarrollar un papel importante en la producción de síntomas cognitivos (córtex prefrontal dorsolateral) y de síntomas afectivos (córtex prefrontal ventromedial) en la esquizofrenia.
- La vía dopaminérgica tuberoinfundibular (d), se proyecta desde el hipotálamo a la glándula hipofisiaria anterior y controla la secreción de prolactina.
- La quinta vía dopaminérgica (e) surge de múltiples sitios, como la sustancia gris periacueductal, el mesencéfalo ventral, de núcleos hipotalámicos y del núcleo parabraquial lateral y desde estos, se proyecta al tálamo. Su función no se conoce bien en la actualidad.
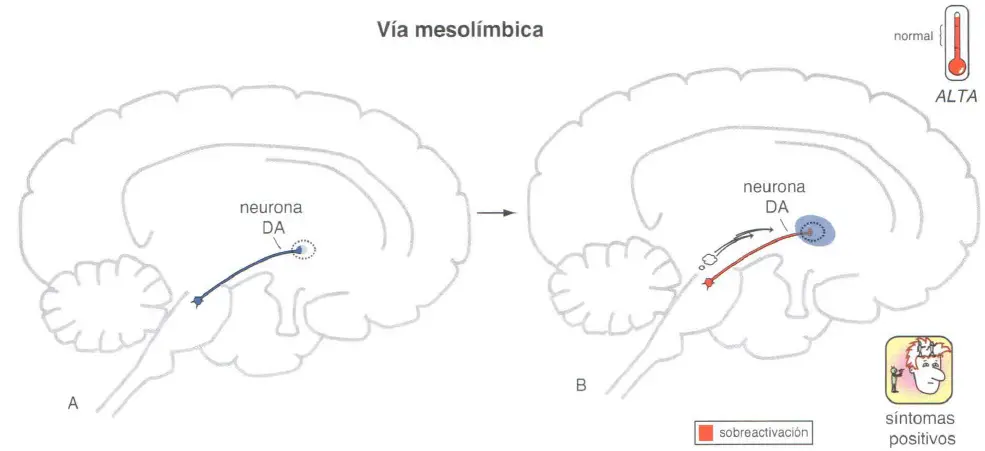
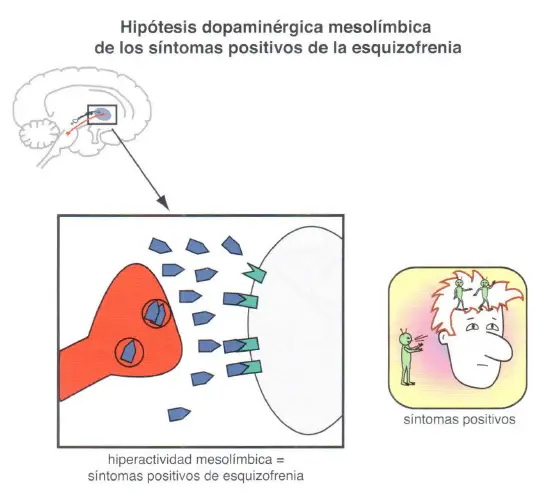
Hipótesis dopaminérgica de la esquizofrenia: la vía dopaminérgica mesolímbica y los síntomas positivos en la esquizofrenia
Durante más de 40 años, se ha observado que las enfermedades o sustancias que aumentan la dopamina potenciarán o producirán síntomas psicóticos positivos, mientras que las sustancias que disminuyen la dopamina atenuarán o eliminarán los síntomas positivos. Por ejemplo, las drogas estimulantes como las anfetaminas o la cocaína liberan dopamina y, si se consumen repetidamente, pueden causar psicosis paranoides virtualmente indistinguibles de los síntomas positivos presentes en la esquizofrenia.
Todos los fármacos antipsicóticos conocidos capaces de tratar los síntomas positivos de las psicosis son bloqueadores del receptor D2 de dopamina. Estas observaciones han sido formuladas en una teoría conocida como «hipótesis dopaminérgica de la esquizofrenia». Quizá una denominación más moderna sería «hipótesis dopaminérgico-mesolímbica de los síntomas positivos de la esquizofrenia», dado que se sabe que es específicamente la hiperactividad en esta vía dopaminérgica la que media la producción de los síntomas positivos en la psicosis.
Teóricamente, la hiperactividad de la vía dopaminérgica mesolímbica explicaría la producción de síntomas positivos en la psicosis, ya sean estos síntomas parte de la enfermedad de la esquizofrenia, de psicosis inducidas por sustancias o síntomas positivos que acompañan a una manía, depresión o demencia. La hiperactividad de las neuronas dopaminérgicas mesolímbicas puede, además tener un papel importante en los síntomas de agresividad y hostilidad en la esquizofrenia y enfermedades relacionadas, especialmente si el control serotoninérgico de la dopamina es deficitario en pacientes que presentan falta de control de los impulsos. Aunque se desconoce qué es lo que produce esta hiperactividad de la dopamina, las teorías actuales afirman que se trata de la consecuencia de una disfunción en el córtex prefrontal y en la actividad glutamatérgica hipocampal.
Vías dopaminérgicas mesocorticales y síntomas cognitivos, negativos y afectivos de la esquizofrenia
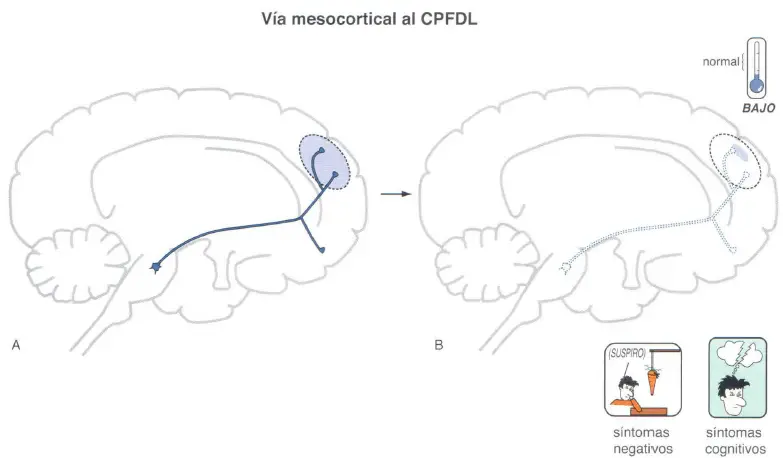
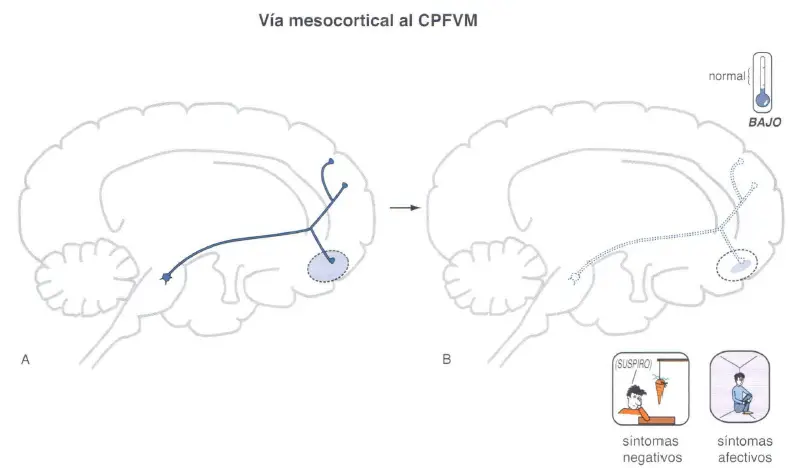
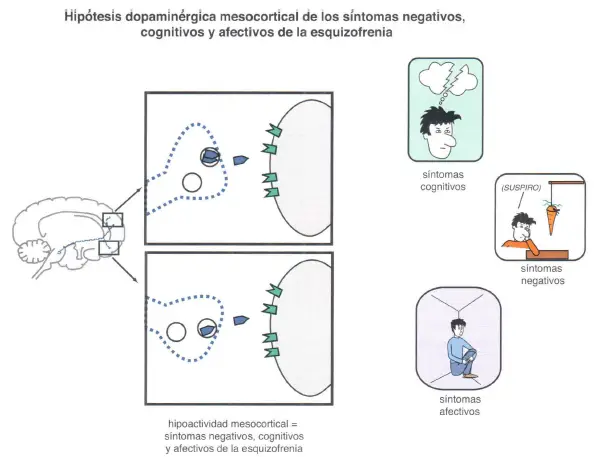
En teoría, el incremento de dopamina en la vía dopaminérgica mesocortical podría mejorar los síntomas negativos, cognitivos y afectivos de la esquizofrenia. Sin embargo, un hipotético exceso de dopamina en otras áreas del cerebro, como la vía mesolímbica, haría que empeorasen los síntomas positivos. Este estado de cosas para la actividad dopaminérgica en el cerebro de los pacientes esquizofrénicos plantea un dilema terapéutico: ¿Cómo aumentar la dopamina en la vía mesocortical mientras, al mismo tiempo, se disminuye la actividad dopaminérgica en la vía mesolímbica?
Vía dopaminérgica mesolímbica, recompensa y síntomas negativos
Cuando un paciente con esquizofrenia pierde motivación e interés y tiene anhedonia y dificultad para experimentar placer, tales síntomas podrían también implicar un funcionamiento deficiente en la vía dopaminérgica mesolímbica, no solo en la vía dopaminérgica mesocortical.
El hecho de que el córtex prefrontal no tenga una alta densidad de receptores D2 podría implicar que exista
un funcionamiento deficiente en el seno del sistema dopaminérgico mesolímbico que provocaría mecanismos de recompensa inadecuados, exhibidos con comportamientos tales como la anhedonia y el abuso de sustancias, así como síntomas negativos como pérdida de la recompensa en las interacciones sociales y disminución global del interés y de la motivación. La incidencia de abuso de sustancias es mucho más elevada en individuos con esquizofrenia que en la población adulta normal, especialmente de nicotina pero también de estimulantes y otras sustancias de abuso; esto podría explicarse en parte como un intento de potenciar una función deficitaria de los centros del placer mesolímbicos, posiblemente a pesar del coste de activar síntomas positivos.
Vía dopaminérgica nigroestriada
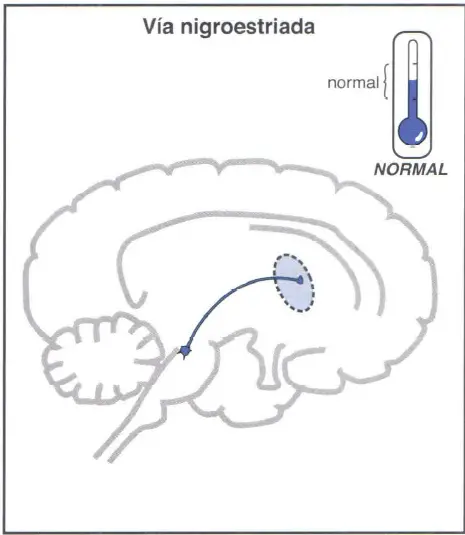
La vía dopaminérgica nigroestriada se proyecta desde la sustancia negra a los ganglios basales o estriado. Es parte del sistema nervioso extrapiramidal y juega un papel clave en la regulación de los movimientos. Cuando existe déficit de dopamina se puede producir parkinsonismo, con temblores, rigidez y quinesia/bradiquinesia. Cuando hay exceso de DA, se producen movimientos hiperquinésicos, como tics o disquinesias. En la esquizofrenia no tratada, se considera que la actividad en esta vía es «normal». El bloqueo crónico de los receptores D2 en esta vía puede provocar un trastorno del movimiento hiperquinésico conocido como disquinesia tardía inducida por neurolépticos.
Vía dopaminérgica tuberoinfundibular
La vía dopaminérgica tuberoinfundibular, del hipotálamo la hipófisis anterior, regula la secreción de prolactina a la circulación. La dopamina inhibe la secreción de prolactina. En la esquizofrenia no tratada, se considera que la actividad de esta vía es «normal». La elevación de los niveles de prolactina se asocia a galactorrea (secreción mamaria), amenorrea (pérdida de la ovulación y de la menstruación) y posiblemente a otros problemas, como disfunción sexual. Estos problemas también pueden ocurrir al recibir tratamiento con los numerosos fármacos antipsicóticos que bloquean los receptores D2.
Vía dopaminérgica talámica
Recientemente, ha sido descrita una vía dopaminérgica
que inerva el tálamo en primates. Se origina en múltiples sitios, incluyendo la sustancia gris periacueductal, el mesencéfalo ventral, diversos núcleos hipotalámicos y el núcleo parabraquial lateral. Su función está todavía en investigación pero puede estar relacionada con el sueño y los mecanismos de mantenimiento de la vigilia, distribuyendo información a través del tálamo al córtex y otras estructuras cerebrales. Actualmente, no hay evidencia de un funcionamiento anormal de esta vía dopaminérgica en la esquizofrenia.
Glutamato
En los últimos años, el neurotransmisor glutamato ha alcanzado un importante papel a nivel teórico en la fisiopatología de la esquizofrenia. El glutamato es el neurotransmisor excitador más importante del sistema nervioso central, considerándose a veces el «interruptor general» del cerebro, ya que es capaz de excitar y encender virtualmente todas las neuronas del SNC. Así, la síntesis, metabolismo, regulación de receptores y las vías clave del glutamato son críticas para el correcto funcionamiento del cerebro y serán revisadas aquí.
Síntesis del glutamato
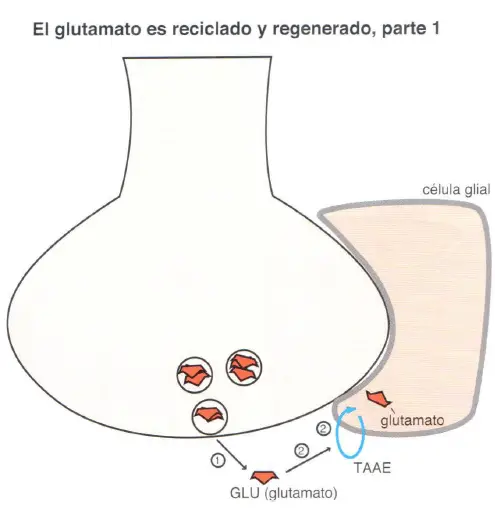
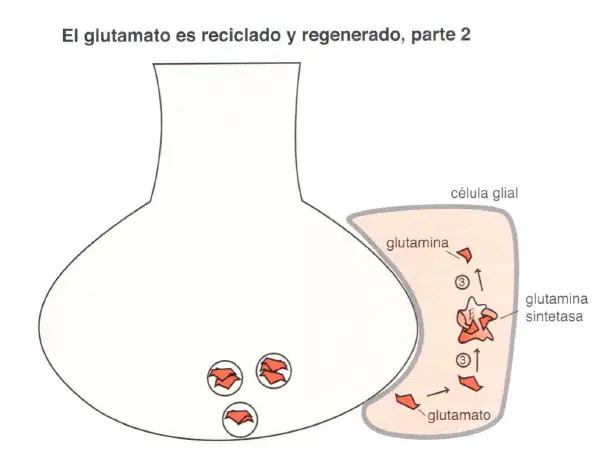
El glutamato o ácido glutámico es un neurotransmisor que es un aminoácido. Su principal uso no es como neurotransmisor sino como aminoácido para la biosíntesis de proteínas. Cuando es usado como neurotransmisor, es sintetizado a partir de la glutamina por las células de la glía, que además ayudan al reciclaje y regeneración de más glutamato tras la liberación del mismo durante la neurotransmisión. Cuando el glutamato es liberado desde las vesículas sinápticas almacenadas en las neuronas glutamatérgicas, interactúa con los receptores de la sinapsis y después es bombeado al interior de las células gliales circundantes mediante una bomba de recaptación llamada transportador de aminoácidos excitadores (TAAE).
 |  |
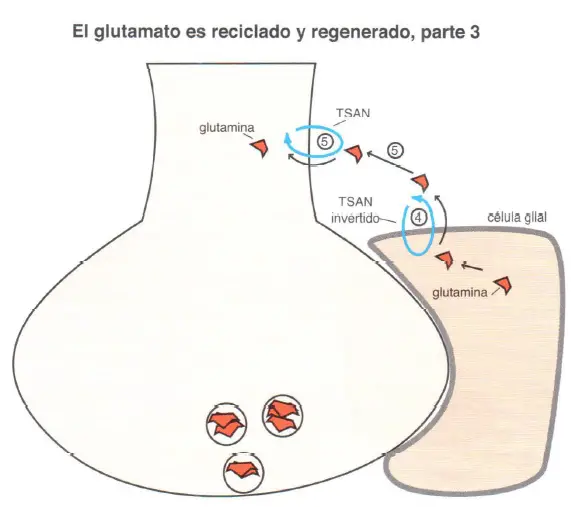 | 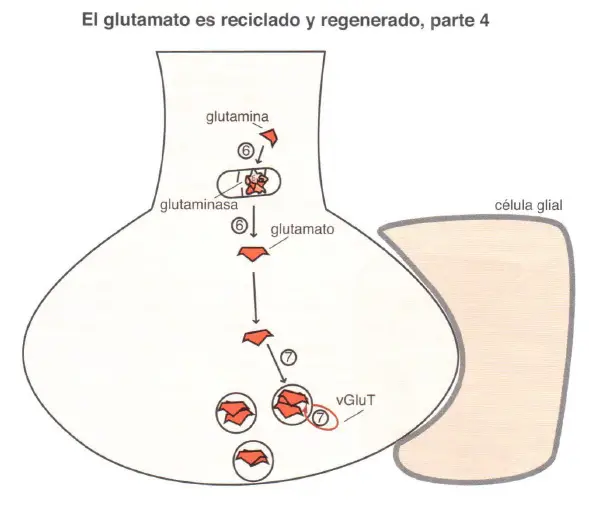 |
Después de la liberación de glutamato desde la neurona presináptica (1 ), este es bombeado al interior de la célula glial vía TAAE, o transportador de aminoácidos excitadores (2). Una vez en el interior de la célula glial, el glutamato es convertido en glutamina por la enzima glutamina sintetasa (3). La glutamina es liberada desde la célula glial por un transportador específico de aminoácidos neutros (TSAN glial) a través de un proceso de transporte inverso (4), y después bombeada por TSANs a la neurona glutamatérgica (5). La glutamina es convertida en glutamato dentro de la neurona presináptica glutamatérgica mediante la enzima glutaminasa (6) y bombeada al interior de las vesículas sinápticas mediante el transportador vesicular de glutamato (vGluT), donde se almacena para futuras liberaciones (7).
Síntesis de cotransmisores del glutamato glicina y D-serina
Los sistemas glutamatérgicos tienen la peculiaridad de que uno de los receptores clave para el glutamato requiere de un cotransmisor, además del glutamato, para funcionar. Ese receptor es el receptor NMDA (Nmetil- o-aspartato ), y el cotransmisor es el aminoácido glicina, o bien otro aminoácido cercano a la glicina, la o-serina.
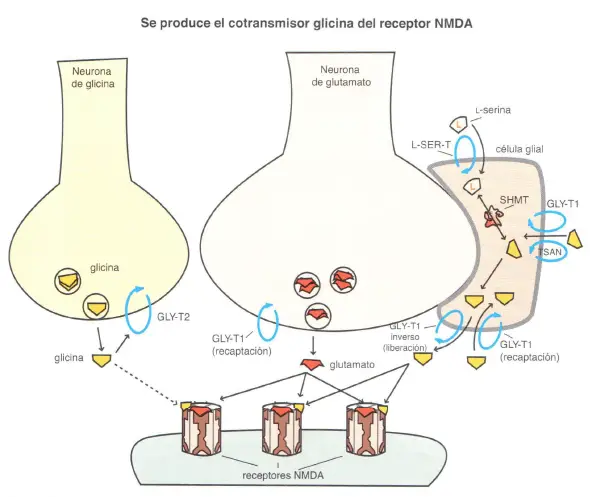
Las acciones del glutamato en el receptor NMDA dependen en parte de la presencia de un cotransmisor, ya sea glicina o serina. La glicina puede proceder directamente de aminoácidos de la dieta y es transportada al interior de la célula glial por un transportador de glicina (Gly-Tl) o por un transportador específico de aminoácidos neutros (TSAN). La glicina además puede ser producida tanto en las neuronas como en las células gliales. Las neuronas productoras de glicina aportan solo una pequeña parte de la glicina presente en las sinapsis glutamatérgicas, porque la mayor parte de la glicina liberada por estas neuronas es usada solamente en las sinapsis de glicina y después recaptada al interior de la presinapsis de la neurona productora de glicina vía el transportador de glicina 2 (Gly-T2), antes de que se pueda difundir mucha glicina a las sinapsis glutamatérgicas. La glicina producida por las células gliales desempeña un papel más amplio en las sinapsis glutamatérgicas. La glicina es producida en la célula glial a partir del aminoácido L-serina, captado por la célula glial vía el transportador de L-serina (L-SER-T), y después es convertida en glicina mediante la enzima serin-hidroximetiltransferasa (SHMT). La glicina de la célula glial es liberada al interior de las sinapsis glutamatérgicas a través de un mecanismo de transporte inverso mediante el transportador de glicina 1 (Gly-Tl ). La glicina extracelular es recaptada por la célula glial por medio de una bomba de recaptación, a saber Gly-Tl.
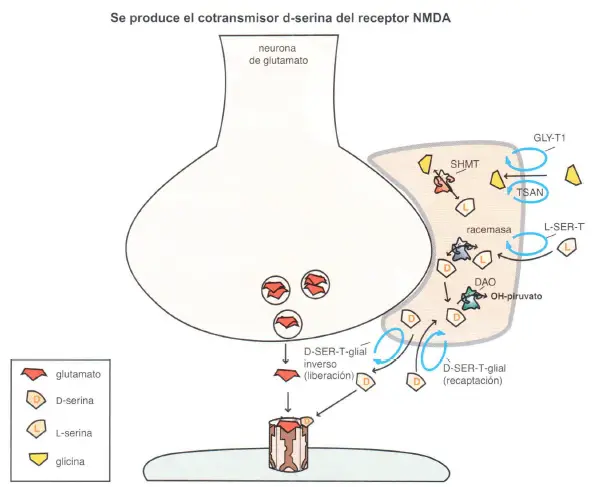
El glutamato requiere la presencia de glicina o serina en el receptor N-metil-o-aspartato (NMDA) para ejercer sus efectos allí. En las células gliales, la enzima serina racemasa transforma la L-serina en o-serina, que a continuación es liberada al interior de la sinapsis glutamatérgica vía transporte inverso gracias al transportador glial o-serina (D-SER-T glial). La célula glial obtiene la L-serina directamente, gracias al transporte mediado por el transportador de L-serina (LSER- T), o por la conversión de glicina en L-serina, gracias a la enzima serin hidroximetil transferasa (SHMT). Una vez que la o-serina es liberada a la sinapsis, es recuperada al interior de la célula glial mediante una bomba de recaptación, denominada D-SER-T El exceso de o-serina en el interior de la célula glial puede ser destruido por la enzima o-aminoácido oxidasa (DAO), que convierte o-serina en hidroxipiruvato (OH-piruvato).
Receptores de glutamato
Hay varios tipos de receptores glutamatérgicos, incluyendo la bomba de recaptación presináptica neuronal (transportador de aminoácidos excitadores, o TAAE) y el transportador de glutamato en las vesículas sinápticas (vGluT).
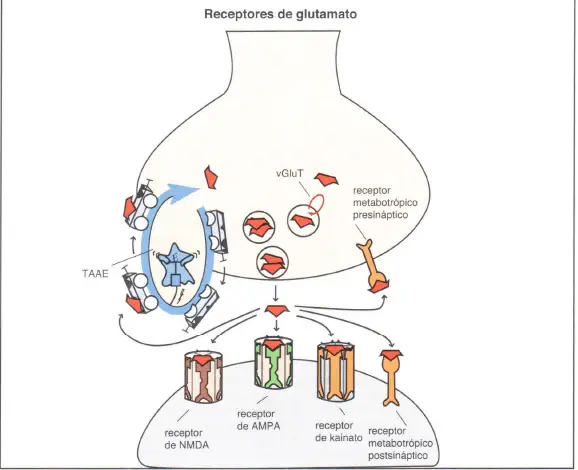
Aquí se muestran los receptores para el glutamato que regulan su neurotransmisión. El transportador de aminoácidos excitadores (TAAE) se localiza en la presinapsis y es responsable de eliminar el exceso de glutamato de la sinapsis. El transportador vesicular (v-Glu-T) introduce glutamato en las vesículas sinápticas, donde se almacena para ser usado en futuras neurotransmisiones. Los receptores metabotrópicos de glutamato (acoplados a proteína G) pueden encontrarse en la pre- o postsinapsis. Tres tipos de receptores de glutamato postsinápticos están acoplados a los canales iónicos, y son conocidos como canales iónicos regulados por ligando: receptores de N-metil-D-aspartato (NMDA), receptores de ácido alfa-amino-3-hidroxi-5-metil-4-isoxazolpropiónico (AMPA) y receptores de kainato, todos denominados por el agonista que se acopla a ellos.
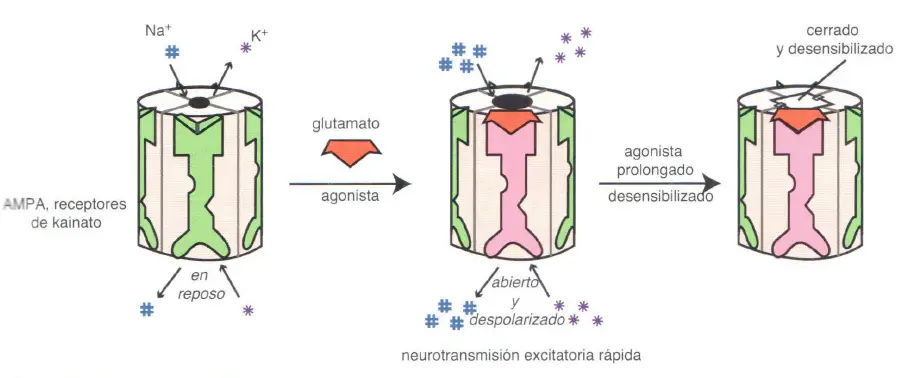
A diferencia de los receptores NMDA, los receptores AMPA y kainato solo necesitan glutamato para unirse y hacer que se abra el canal. Esto conduce a una neurotransmisión excitatoria rápida y una despolarización de membrana. La unión mantenida del agonista glutamato dará lugar a una desensibilización del receptor, haciendo que se cierre el canal y que, transitoriamente, deje de tener respuesta al agonista.
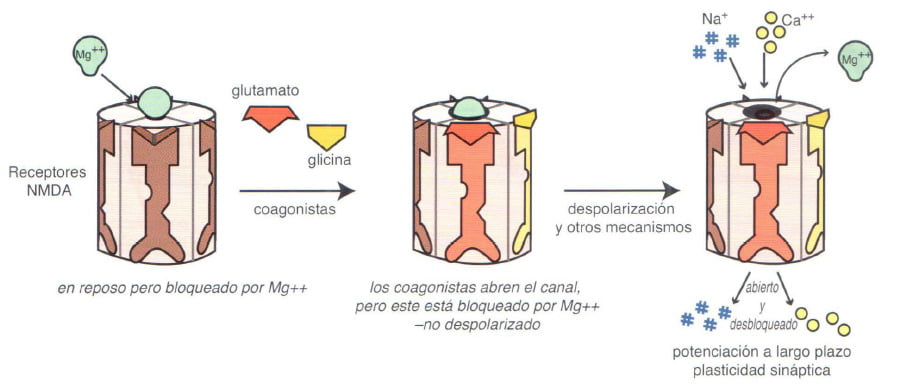
El magnesio es un modulador alostérico negativo (NAM) en los receptores glutamatérgicos NMDA. La apertura de los receptores glutamatérgicos NMDA requiere la presencia de glutamato y glicina, uniéndose cada uno de ellos a una zona diferente del receptor. Cuando el magnesio también está unido y la membrana no está despolarizada, se impiden los efectos de glutamato y glicina y no se permite la apertura del canal iónico. Para que el canal se abra, la despolarización debe retirar el magnesio mientras que glutamato y glicina están unidos a sus zonas de unión en el complejo de cana iónico regulado por ligando.
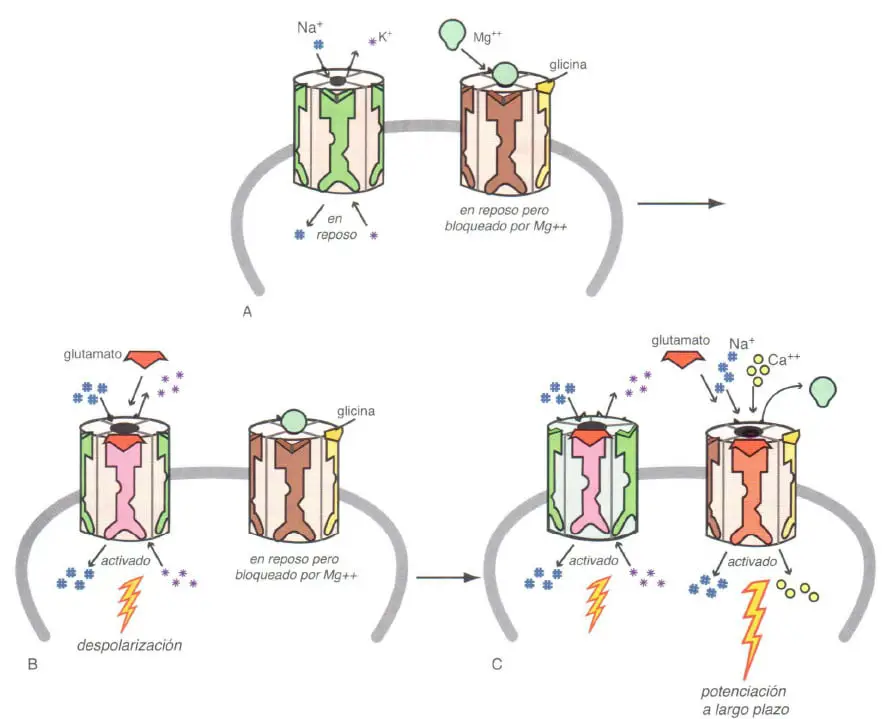
(A) A la izquierda hay un receptor AMPA con su canal de sodio en estado de reposo, lo que permite la entrada de una mínima cantidad de sodio en la célula a cambio de potasio. A la derecha hay un receptor NMDA con magnesio que bloquea el canal de calcio y glicina unida a su lugar de unión. (B) Cuando llega el glutamato, se une al receptor AMPA, haciendo que el canal de sodio se abra, aumentando así el flujo de sodio hacia dentro de la dendrita y de potasio hacia fuera. Esto hace que la membrana se despolarice y activa un impulso nervioso postsináptico. (C) La despolarización de la membrana retira el magnesio del canal de calcio. Esto, unido a la unión del glutamato al receptor NMDA en presencia de glicina, hace que se abra el receptor y permite el influjo de calcio. El influjo de calcio a través de los receptores NMDA contribuye a la potenciación a largo plazo, un fenómeno que podría intervenir en el aprendizaje a largo plazo, la sinaptogénesis y otras funciones neuronales.
TABLA RECEPTORES DE GLUTAMATO
Receptores metabotrópicos acoplados a proteínas G. Existen 8 subtipos organizados en 3 grupos ( II y III)
| GRUPO I | GRUPO II | GRUPO III | |
| mGluR1 mGluR5 | mGluR2 mGluR3 | mGluR4 mGluR6 mGluR7 mGluR8 | |
| POSTSINÁPTICOS (facilitan y potencian la neurotransmisión excitadora glutamatérgica) | X | ||
| PRESINÁPTICOS ( bloquean/reducen la liberación de glutamato) | X | X |
lonotrópicos (canales iónicos regulados por ligando; receptores acoplados a canal iónico)
| Clase funcional | Familia genética | Agonistas | Antagonistas |
| AMPA | GluR1 GluR2 GluR3 GluR4 | Glutamato AMPA Kainato | |
| Kainato | GluR5 GluR6 GluR7 KA1 KA2 | Glutamato Kainato | |
| NMDA | NR1 NR2A NR2B NR2C NR2D | Glutamato Aspartato NMDA | MK801 Quetamina PCP (fenciclidina) |
Principales vías glutamatérgicas del cerebro
El glutamato es un neurotransmisor excitatorio ubicuo que parece ser capaz de excitar casi a cualquier neurona del cerebro; por este motivo, en algunas ocasiones, se lo denomina «interruptor general». Además, hay una media docena de vías glutamatérgicas específicas que tienen especial relevancia en psicofarmacología y especialmente para la fisiopatología de la esquizofrenia.
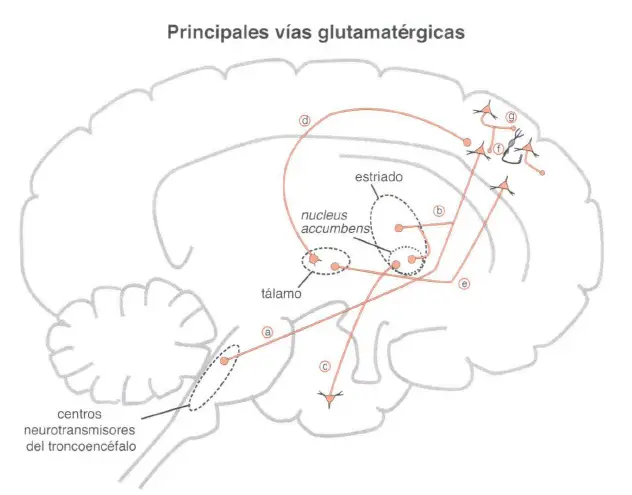
Aunque el glutamato puede actuar virtualmente sobre cualquier neurona del cerebro, hay cinco vías glutamatérgicas particularmente relevantes para la esquizofrenia. (a) La proyección glutamatérgica córtico-troncoencefálica es una vía descendente que se proyecta desde las neuronas piramidales del cortex prefrontal a centros neurotransmisores del troncoencéfalo (rafe, locus coeruleus, área tegmental ventral, sustancia negra) y regula la liberación de neurotransmisores. (b) Otra vía descendente del glutamato se proyecta del córtex prefrontal al estriado (vía glutamatérgica corticoestriada) y al nucleus accumbens (vía glutamatérgica córtico-accumbens}, y constituye la porción «córticoestriada» de los haces córticoestriado-talámicos. (c) Vía hipocampal-accumbens (del hipocampo ventral al núcleo accumbens); (d) Vía tálamo-cortical (del tálamo al córtex prefrontal); (e) Vía cortico-talámica (del córtex prefrontal al tálamo); (f} Vía cortico-cortical (neuronas intracorticales que comunican neuronas glutamatérgicas entre sí, dentro de la corteza); (g) Neuronas intracorticales, que comunican células glutamatérgicas de la corteza, vía interneuronas GABAérgicas.
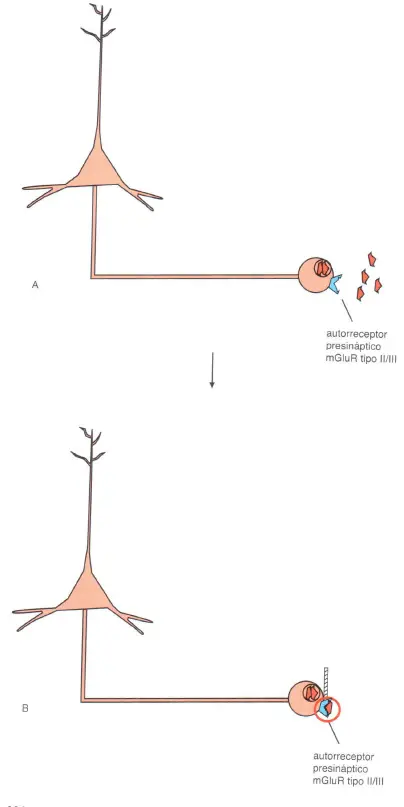
Los receptores glutamatérgicos metabotrópicos de los Grupos II y III pueden existir presinápticamente como autorreceptores para regular la liberación de glutamato. Cuando se elabora el glutamato en la sinapsis (A), está disponible para unirse alautorreceptor, el cual entonces inhibe la liberación de glutamato (B).
Vías glutamatérgicas córtico-troncoencefálicas (a)
Vía glutamatérgica descendente muy importante, se proyecta desde las neuronas córtico piramidales, a centros de neurotransmisión del troncoencéfalo, incluyendo los núcleos del rafe encargados de la neurotransmisión serotoninérgica, el área tegmental ventral (ATV) y la sustancia negra, de la dopaminérgica, y el locus coeruleus, de la noradrenérgica. Esta vía es clave en la regulación de la liberación de neurotransmisores.
Vías glutamatérgicas corticoestriadas (b)
Vía glutamatérgica descendente de las neuronas piramidales es la que se proyecta al estriado, se proyecta al estriado dorsal, o vía glutamatérgica córtico-accumbens, cuando se proyecta a un área específica del estriado ventral conocida como nucleus accumbens. En ambos casos, estas vías glutamatérgicas descendentes terminan en las neuronas GABA destinadas a una estación de relé en otra parte del complejo estriatal denominada globus pallidus.
Vía glutamatérgica hipocampal-accumbens (c)
Vía glutamatérgica principal, se proyecta desde el hipocampo al nucleus accumbens. Existen teorías específicas que relacionan esta vía concreta con la esquizofrenia. Como las vías glutamatérgicas córtico-estriatal y córtico-accumbens, la proyección glutamatérgica hipocampal al nucleus accumbens también termina en las neuronas GABA que, por su parte, se proyectan a una estación de relé en el globus pallidus.
Vía glutamatérgica talamo-cortical (d)
Esta vía lleva información de vuelta desde el tálamo hasta el córtex, a menudo para procesar información sensorial.
Vía glutamatérgica córtico-talámica (e)
Vía glutamatérgica, se proyecta directamente al tálamo, donde podría orquestar la forma en que las neuronas reaccionan a información sensoriales.
Vía glutamatérgica córtico-cortical directa (f)
Complejo de muchas vías glutamatérgicas presentes en el córtex Por un lado, las neuronas piramidales pueden excitarse entre sí en el córtex cerebral por medio de las entradas sinápticas directas desde su propio neurotransmisor glutamato.
Vía glutamatérgica córtico-cortical indirecta (g)
Una neurona piramidal puede inhibir a otra por medio de entradas indirectas, concretamente por medio de interneuronas que liberan GABA.
Hipótesis de la hipofunción del receptor NMDA en la esquizofrenia: quetamina y fenciclidina
Es una de las hipótesis actuales más importantes sobre la causa de la esquizofrenia; propone que la actividad del glutamato en NMDA es hipofuncional debido a anomalías en la formación de las sinapsis de NMDA glutamatérgicos durante el neurodesarrollo. Hipotéticamente, las anomalías genéticas también producen hipofuncionalidad en los receptores NMDA y sus sinapsis originando la propia esquizofrenia. La anfetamina, que libera dopamina, también produce un estado psicótico de delirios y alucinaciones en personas normales similar al de los síntomas positivos de la esquizofrenia. Lo que hace tan atractiva la hipótesis de la hipofunción de los receptores NMDA en la esquizofrenia es que, a diferencia de las anfetaminas que provocan solo síntomas positivos, la PCP además mimetiza los síntomas cognitivos, negativos y afectivos de la esquizofrenia como aislamiento social y disfunción ejecutiva. También puede explicar la hipótesis de la dopamina en esquizofrenia, concretamente, como consecuencia de la hipofunción de los receptores
NMDA.
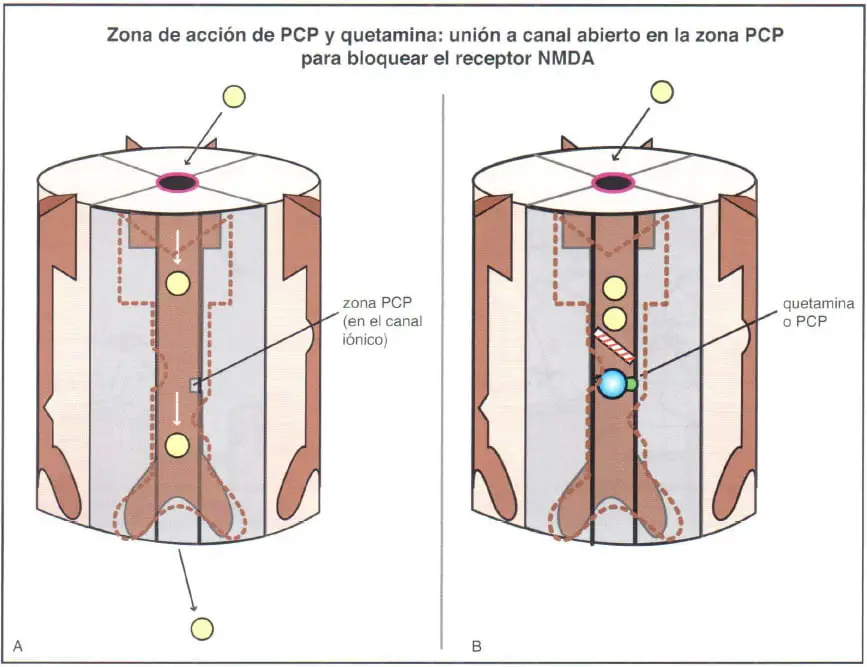
La quetamina anestésica se une a la conformación del canal abierto del receptor N-metil-D-aspartato (NMDA). Concretamente, se une en una zona dentro del canal de calcio de este receptor, que suele denominarse zona PCP porque es también donde la fenciclidina (PCP) se une. El bloqueo de receptores NMDA podría evitar las acciones excitatorias del glutamato.
Hipótesis de la hipofunción de NMDA en la esquizofrenia: sinapsis de NMDA deficiente en interneuronas GABA dentro del córtex prefrontal
Aunque los receptores NMDA y sinapsis están presentes por todo el cerebro y PCP o quetamina los bloquean, existe una teoría actual bien asentada sobre la esquizofrenia que sugiere que la esquizofrenia podría estar causada por anomalías del neurodesarrollo en la formación de las sinapsis glutamatérgicas en una zona específica, concretamente, en ciertas interneuronas GABA del córtex cerebral. Parecería que algo está mal en la programación genética de aquellas interneuronas GABA específicas que pueden ser identificadas en el córtex prefrontal como contenedoras de una proteína de unión de calcio denominada parvalbúmina.
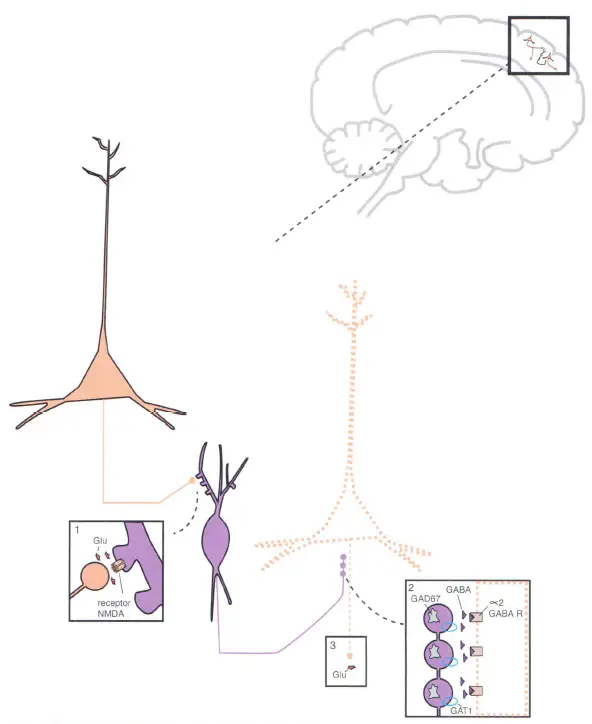
En la imagen se muestra un detalle de las neuronas piramidales corticales que se comunican por medio de interneuronas GABAérgicas. (1) Se libera glutamato desde una neurona piramidal intracortical y se une a un receptor NMDA en una interneurona GABAérgica. (2) Entonces se libera GABA desde la interneurona y se une a receptores GABA del subtipo a2 que están ubicados en el axón de otra neurona piramidal glutamatérgica. (3) Esto inhibe la neurona piramidal, reduciendo así la liberación posterior de glutamato.
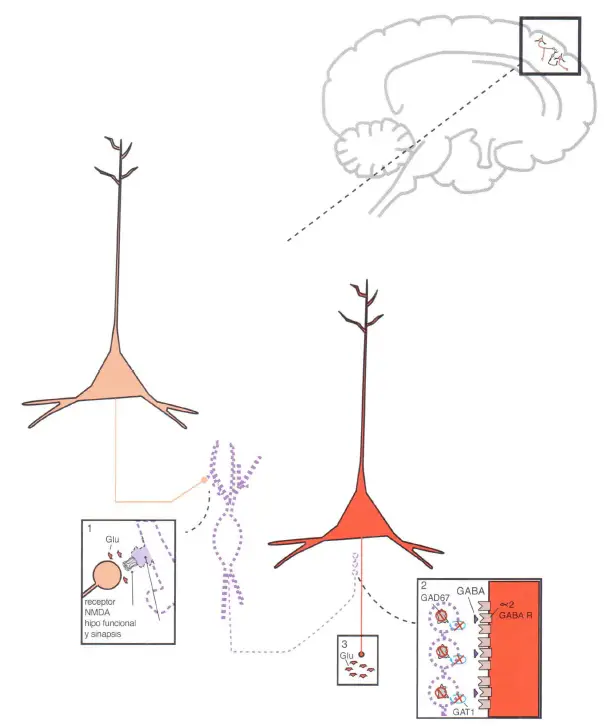
En la imagen se muestra un detalle de las neuronas piramidales corticales comunicándose por medio de interneuronas GABAérgicas en presencia de receptores NMDA hipofuncionales. (1) El glutamato es liberado desde una neurona piramidal intracortical. Sin embargo, el receptor NMDA al que se une es hipofuncional, lo que impide que el glutamato ejerza su efecto pleno vía el receptor NMDA. (2) Esto impide la liberación de GABA desde la interneurona; así, la estimulación de receptores a , GABA en el axón de otra neurona glutamatérgica no tiene lugar. (3) Cuando GABA no se une a los receptores a , GABA en su axón, la neurona piramidal deja se ser inhibida y pasa a estar desinhibida y hiperactiva, liberando una cantidad excesiva de glutamato.
¿Cuáles son las consecuencias de la hipotética desconectividad del glutamato con estas interneuronas GABA en particular? Cuando las interneuronas GABA que contienen parvalbúmina dejan de funcionar adecuadamente, no inhiben correctamente las principales neuronas piramidales glutamatérgicas en el córtex prefrontal, haciendo que esas neuronas de glutamato se vuelvan hiperactivas. Esto interrumpe hipotéticamente el funcionamiento de las neuronas posteriores, especialmente dopamina neuronas. De este modo, una sinapsis enferma en un circuito neuronal puede afectar al conjunto del circuito, desde la interneurona GABA y las neuronas de glutamato que inerva, hasta las neuronas de dopamina posteriores y más allá.
Relación de la hipótesis de la hipofunción NMDA en esquizofrenia con la hipótesis dopaminérgica en esquizofrenia: síntomas positivos
Un complejo conjunto de interacciones permite al glutamato determinar la liberación de dopamina. Las vías glutamatérgicas que regulan las vías dopaminérgicas mesolímbicas y mesocorticales son fundamentales en la esquizofrenia.
(A) Las proyecciones glutamatérgicas córtico-troncoencefálicas se comunican directamente con la vía dopaminérgica mesolímbica en el área tegmental ventral (ATV) para regular la liberación de dopamina en el nucleus accumbens. (B) Si existe hipoactividad de los receptores NMDA en las interneuronas GABA corticales, la vía córtico-troncoencefálica a la ATV estará hiperactivada, dando lugar a una liberación excesiva de glutamato en la ATV. Esto dará lugar a una estimulación excesiva de la vía dopaminérgica mesolímbica, y así a una liberación excesiva de dopamina en el nucleus accumbens. Esta es la base biológica teórica para la hiperactividad de dopamina mesolímbica que estaría asociada con los síntomas positivos de la psicosis.
Los receptores NMDA hipofuncionales en las sinapsis glutamatérgicas del hipocampo ventral también pueden contribuir a la hiperactividad de dopamina mesolímbica. (A) El glutamato liberado en el hipocampo ventral se une al receptor NMDA en una interneurona GABAérgica, estimulando la liberación de GABA. GABA se une a los receptores en una neurona glutamatérgica piramidal que se proyecta al nucleus accumbens; esto inhibe la liberación de glutamato aquí. La relativa ausencia de glutamato en el nucleus accumbens permite una activación normal de la neurona GABAérgica que se proyecta al globus pal/idus, lo que a su vez permite la activación normal de una neurona GABAérgica que se proyecta al ATV. Esto da lugar a una activación normal de la vía dopaminérgica mesolímbica desde el ATV al nucleus accumbens_ (B) Si los receptores NMDA de las interneuronas GABA del área ventral hipocampal son hipoactivos, se sobreactivará entonces la vía glutamatérgica al nucleus accumbens, dando lugar a una liberación excesiva de glutamato en el nucleus accumbens. Esto llevará a la estimulación excesiva de neuronas GABAérgicas que se proyectan al globus pal/idus, lo que a su vez inhibirá la liberación de GABA desde el globus pallidus al ATV. Esto producirá la desinhibición de la via dopaminérgica mesolímbica y así una libración excesiva de dopamina en el nucleus accumbens.
La conclusión es que una producción excesiva de glutamato aguas arriba desde el córtex prefrontal o el hipocampo puede contribuir a la hiperactividad dopaminérgica posterior y a los síntomas positivos de esquizofrenia.
Relación de la hipótesis de hipofunción de NMDA en la esquizofrenia con la hipótesis dopaminérgica en la esquizofrenia: síntomas negativos
Al parecer, las neuronas glutamatérgicas en la vía córtico-troncoencefálica que regulan las neuronas dopaminérgicas del ATV, que se proyectan únicamente al córtex prefrontal (vía dopaminérgica mesocortical), son diferentes a las que regulan las neuronas dopaminérgicas del ATV que se proyectan al nucleus accumbens como a la vía dopaminérgica mesolímbica. Así, diferentes poblaciones de neuronas glutamatérgicas regulan las diferentes poblaciones de neuronas dopaminérgicas.
(A) La proyección de glutamato troncoencefálico cortical se comunica con la vía dopaminérgica mesocortical en el área ventral tegmental (ATV) via nterneuronas GABA inhibitorias, regulando así la liberación de dopamina en el córtex prefrontal. (B) Si los receptores NMDA de las interneuronas GABA corticales son hipoactivos, entonces la vía córtico-troncoencefálica al ATV quedará hiperactivada, dando lugar a una liberación excesiva de glutamato en el ATV. Esto dará lugar a una estimulación excesiva de las interneuronas GABAérgicas del tallo cerebral, que a su vez producirá la inhibición de las neuronas dopaminérgicas mesocorticales. Esto reduce la liberación de dopamina en el córtex prefrontal y es la base biológica teórica de los síntomas negativos de psicosis.
En el manual se dice que cuando el glutamato es usado como neurotransmisor, es sintetizado a partir de la glutamina por las células de la glía. Pero, por lo que se describe en el resto del texto, parece que en realidad el glutamato se sintetiza únicamente en la neurona a partir de la glutamina, mediante la enzima llamada glutaminasa, limitándose la célula glial al reciclado y regeneración del glutamato, siendo únicamente capaz de realizar el proceso inverso, es decir, de convertir el glutamato en glutamina mediante la glutamina sintetasa. ¿Es realmente así?
La psicofarmacología es una disciplina muy dinámica y lo que hoy es ley-teoría probada, mañana se cuestiona y es sustituida por otra novedosa empíricamente comprobada. Es lo que sucede en este caso específico, en donde la glutamina es el precursor del glutamato u origen de éste en la neurona, mediante reacciones anapleróticas (de ‘relleno’, o sea completando, en el proceso de oxidación –metabolismo oxidativo- de lípidos, grasas, aminoácidos, etc.). Las reacciones anapleróticas (término del griego, ‘relleno’) son aquellas que proporcionan intermediarios del ciclo de los ácidos tricarboxílicos (TCA, del inglés) o ciclo del ácido cítrico o ciclo de Krebs (estudiado en el primer curso del Grado).
En realidad, y aunque en el texto y gráficos se pueda dar a entender que la síntesis del glutamato sólo se produce en la neurona no parece ser del todo cierto; sino más bien, todo lo contrario: [http://www.madrimasd.org/blogs/biocienciatecnologia/2010/09/25/131650]
De hecho, recientes investigaciones llevadas a cabo por un equipo español del CBMSO (Centro de Biología Molecular Severo Ochoa de Madrid del CSIC-UAM ubican la síntesis ex novo de glutamato en las células gliales (astrocitos) a partir del aminoácido precursor aspartato (pdf. adjunto) que pasa precisamente desde la neurona a la célula glial, donde comienza de nuevo el proceso de síntesis de glutamato a partir de aquél en el astrocito; y de transformación en la propia neurona (desde la glutamina a través de la glutaminasa).
Vamos a aclararlo:
Parece ser que hay dos vías en el origen del glutamato: 1) el que procede de la neurona , mediante la transformación -a través del enzima glutaminasa (fig. 4.19D)- de la glutamina cuya fuente procede de las células gliales en donde se sintetiza aquél (la glutamina, aminoácido excitador) mediante la actividad de la glutamina sintetasa (Fig. 4.19B); 2) la otra vía de generación del glutamato es una vía de intercambio transcelular en un ciclo en el que intervienen, como se ha mencionado anteriormente, una síntesis nueva de glutamato y glutamina a partir de la presencia y activación del –intermediario- aspartato mediante el proceso de síntesis anaplerótica que se produce en el propio astrocito. 3) También, por supuesto, en la célula glial se produce no sólo la génesis señalada (regeneración del transmisor glutamato a partir del aspartato y glutamina), sino el reciclaje de éste para volver a ser reutilizado tras ser liberado por el terminal neuronal durante la neurotransmisión, transformándose a su vez -en el interior de la célula glial- en glutamina por la glutamina sintetasa (pág. 96); esta última vía sería, por supuesto, de aporte de nueva glutamina (reciclada y creada) para completar la vía 1 en la neurona aumentando los depósitos necesarios para la transformación en más moléculas de glutamato necesarios para el paso subsecuente de creación/génesis de la vía 2. Se cierra el círculo. De momento…
Tal como indican los autores del trabajo (), descubridores del elemento del grupo amino necesario para la síntesis de neurotransmisor glutamato, se requiere un ciclo de intercambio transcelular para la síntesis de glutamato y cuyo origen es considerado en la actualidad el astrocito, que ha pasado a tener un papel primordial en la modulación y regulación de la neurotransmisión y no sólo en el reciclaje y sustento estructural neuronal como hasta hace poco tiempo era considerada la glía. () «The glutamate–glutamine cycle faces a drain of glutamate by oxidation, which is balanced by the anaplerotic synthesis of glutamate and glutamine in astrocytes. De novo synthesis of glutamate by astrocytes requires an amino group whose origin is unknown. We find that aspartate, but not other amino acids, increases glutamate synthesis in both control and aralar-deficient astrocytes, mainly by serving as amino donor. …These findings suggest the existence of a neuron-to-astrocyte aspartate transcellular pathway required for astrocyte glutamate synthesis and subsequent glutamine formation. This pathway may provide a mechanism to transfer neuronal-born redox equivalents to mitochondria in astrocytes» (Pardo et al., 2011).
AUTOEVALUACIÓN
Referencias
Stahl, S., & Muntner, N. (2016). Psicofarmacología esencial de Stahl : Bases neurocientíficas y aplicaciones prácticas (4ª ed.; Ed. especial para alumnos de la UNED. ed., Aula Médica Formación en Salud). Madrid: Aula Médica.
YouTube
