

Versión 4
INTRODUCCIÓN

En 1866, John Langdon Down, describió de forma sistemática un tipo de discapacidad intelectual integrado por «idiotas congénitos» con las características físicas que se habían establecido como propias de la raza mongólica. Además de enumerar los rasgos faciales patognomónicos (cara ancha, ojos oblicuos, lengua gruesa, nariz pequeña), Down recogió otros aspectos del cuadro, como la capacidad de imitación, la voz gruesa y confusa, o las dificultades de coordinación. El término «mongolismo» fue utilizado para describir esta alteración hasta que, en la década de 1960, el grupo de Lejeune identifica su base genética y se decide sustituir por un término menos ofensivo como «trisomía 21 o síndrome de Down» (Martínez Pe?rez, 2011).
Recuadro 9-1. Genética del Síndrome de Down
El Síndrome de Down es una alteración cromosómica caracterizada por un exceso de material cromosómico en el par 21, con tres variantes conocidas: trisomía 21 primaria, trisomi?a por translocacio?n y trisomi?a por mosaico:
- La Trisomía es la alteración más frecuente (90-95%). El cariotipo muestra una copia de más en el par de cromosomas 21. En 1991 Petersen y Cols. demostraron que sólo la triplicación de la banda 21q22 del cromosoma es suficiente para que se manifieste el síndrome.
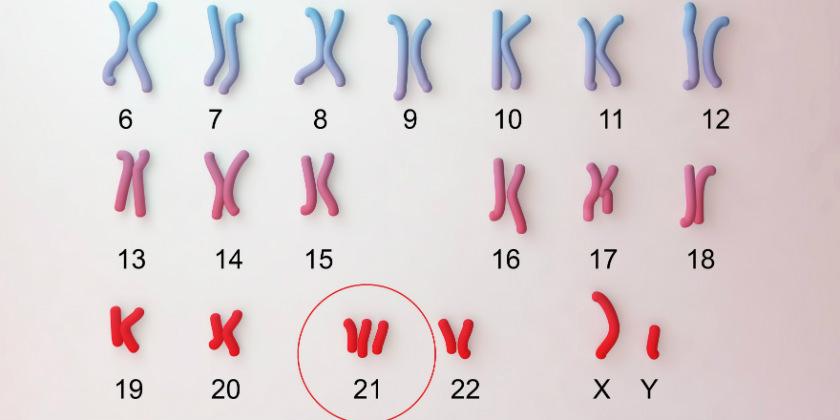
- En la Translocación, el cariotipo presenta sólo 46 cromosomas, pero en uno de los miembros de un par de cromosomas, p. ej., del 14 o del 15, se detecta un brazo más largo; al analizarlo se comprueba que es material genético del cromosoma 21 y, de hecho, la persona puede exhibir características similares a las de la trisomía.
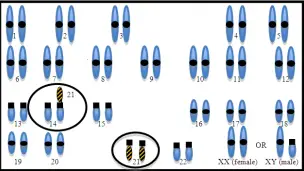
- En el Mosaicismo puede ocurrir una de las dos opciones siguientes:
- Que el cigoto haya recibido 46 cromosomas pero que, tras un error en la primera división celular, los cromosomas del par 21 no se hayan separado bien, por lo que una célula recibirá 45 (no será viable) y otra 47. Ésta dará lugar a una línea celular con un cromosoma 21 de más. El resultado final será que el individuo podrá tener en su organismo unas células con 46 cromosomas y otras con 47.
- Que el cigoto haya recibido 47 cromosomas desde su inicio (ej. de un progenitor portador de translocación), y en sucesivas divisiones algunas células hayan perdido el cromosoma 21 extra y otras no.
La variabilidad sintomática del mosaicismo es mayor, dado que el fenotipo dependerá de cuántos y cuáles sean los órganos afectados por la presencia de células trisómicas: habrá niños sin los rasgos faciales típicos del síndrome, quienes presenten cardiopatías y quienes no. Lo mismo ocurrirá con las capacidades intelectuales y los déficits neuropsicológicos. Por lo general, el perfil neuropsicológico asociado al mosaicismo suele ser mejor que el de los afectados por trisomía o translocación.
«No obstante, si le interesa ampliar la información sobre este tema le recomiendo que visite la página web: www.mosaicdownsyndrome.com, en ella podrá encontrar explicación más detallada como ésta que le adjunto aquí mismo, por si otros alumnos pudieran estar interesados también en ello, a pesar de exceder los contenidos básicos, que se les pide conocer para este curso de Neuropsicología del Desarrollo». Docente Maria Angeles Perez
«Síndrome de Down Mosaicismo
Dentro de las formas que adopta la trisomía 21 o síndrome de Down, hay una denominada “mosaico” que afecta aproximadamente al 1-1,5 % de las personas que tiene este diagnóstico. Su presencia suele despertar una gran intranquilidad en los padres. En primer lugar porque, al presentarse con una frecuencia tan pequeña, existe un menor conocimiento de las características con que se expresa en un niño determinado; y en segundo lugar, porque existe una opinión generalizada que se transmite de unos a otros, de que en las personas con síndrome de Down mosaico el retraso mental o discapacidad intelectual será más leve.
En este artículo vamos a exponer los datos más fundamentales que conocemos sobre el síndrome de Down mosaico, y ofreceremos algunas orientaciones prácticas que sirven a los padres para afrontar con cierta seguridad la crianza y la educación de su hijo.
Síndrome de Down Mosaico
A QUÉ LLAMAMOS MOSAICISMO
El término mosaicismo no es exclusivo del síndrome de Down. Mosaicismo –palabra tomada del término mosaico en el que el conjunto artístico está formado por piezas o materiales de varios colores– indica que una persona posee una composición cromosómica en sus células que no es homogénea sino que presenta variantes. En el síndrome de Down, concretamente, algunas células del cuerpo tienen 46 cromosomas, que es el número normal, mientras que otras células tienen 47 cromosomas, y ese cromosoma extra pertenece a la pareja 21 por lo que presentan trisomía 21.
¿A QUÉ SE DEBE EL MOSAICISMO?
Recordemos que cada organismo humano proviene de una primera célula inicial, llamada zigoto, que se origina tras la fecundación del óvulo femenino por parte del espermatozoide masculino. Cada uno de ellos aporta 23 cromosomas que, al sumarse, conforman los 46 cromosomas del zigoto. A partir de esta primera célula, se sucede una serie ininterrumpida de divisiones celulares (cada célula se divide en otras dos) que incluye la división de sus cromosomas en dos mitades (en sentido longitudinal). Las células que se originan van emigrando a diversos sitios y van diferenciándose, es decir, adoptando formas y funciones distintas para originar los diversos tejidos, órganos, aparatos y sistemas. En condiciones normales, todas esas células mantienen en su núcleo 46 cromosomas.
Ahora bien, en ocasiones una de las primeras divisiones celulares puede cometer un error en la división y separación de sus cromosomas, de modo que una de las células divididas se hace con un número distinto de cromosomas (por ejemplo, 47 en lugar de 46). Todas las células que se originen a partir de esa célula (las cuales constituyen lo que se llama una “línea o linaje celular”)
Seguirán teniendo ese número irregular o anómalo de cromosomas mientras que las demás tendrán el número normal de 46. Al final el organismo ya formado tendrá células con 46 cromosomas y células con 47.
Lógicamente, cuanto más tempranamente haya aparecido esa anomalía en el curso de la división celular (es decir, en las primeras “generaciones” de células), más probable será que el número final de células que contengan anomalía sea alto. A eso llamamos porcentaje de mosaicismo: 10 % significa que sólo el 10 % de las células del organismo posee el número anómalo de cromosomas y el 90 % de las células posee el número normal.
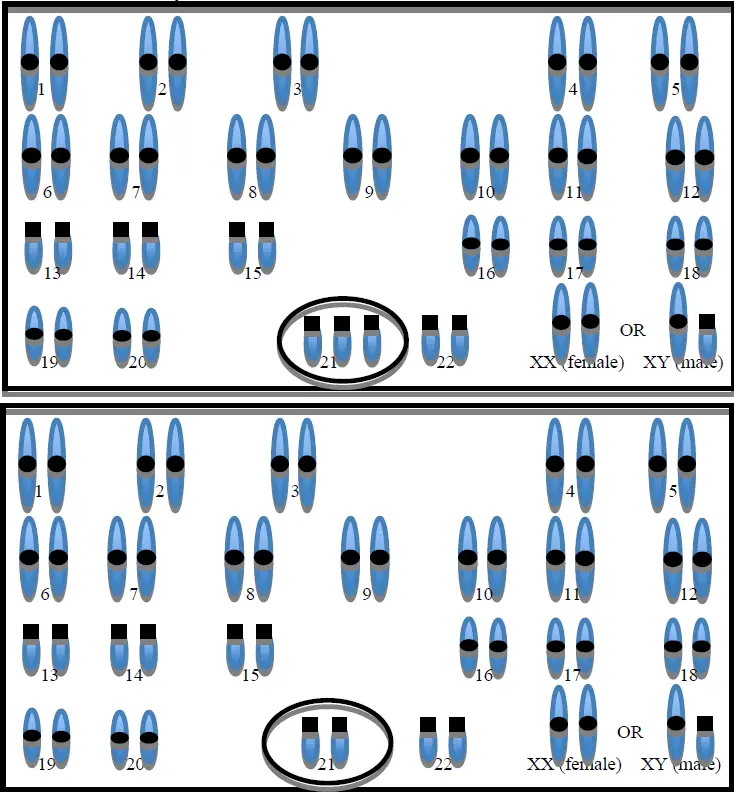
En el caso del síndrome de Down, la anomalía de la división y separación cromosómica queda restringida a la pareja 21; de modo que, a partir de una célula de las muchas ya formadas, con sus 2 cromosomas 21, se forman se forman dos células de las que una contiene tres cromosomas 21 y la otra uno solo (esta última no suele ser viable, es decir, no se vuelve a reproducir y muere) (fig. 1).
Existe otra manera de formarse el mosaicismo 21. En ocasiones, es el zigoto el que ya tiene los tres cromosomas 21 propios del trisomía. Pero en el curso de la división celular, una o más de las líneas celulares pierden el tercer cromosoma 21, quedándose con el número normal.
DIAGNÓSTICO DEL MOSAICISMO
El síndrome de Down o trisomía 21 se diagnostica mediante el estudio del cariotipo, es decir de los cromosomas presentes en las células. Habitualmente se realiza mediante la toma de sangre y análisis de varias células sanguíneas: 20 o 25. En el caso de que todas ellas posean 47 cromosomas 21 hacemos el diagnóstico de trisomía simple, pero si algunas tienen 47 y otras 46, hacemos el diagnóstico de síndrome de Down mosaico. El diagnóstico queda hecho, pero no olvidemos que en la sangre sólo existen células de la “línea celular sanguínea”, por lo que el análisis de sangre sólo revela el mosaicismo en esa línea celular. ¿Habrá también en otras?
Es posible que la división celular en mosaico se haya dado en otras líneas celulares presentes en otros tejidos y órganos; por ejemplo, en la piel. Por ese motivo, si hay sospecha de mosaicismo en el análisis de sangre es conveniente realizar el cariotipo también en otros tejidos, siendo los más asequibles la piel y la médula ósea. La ventaja de hacerlo en la piel estriba en el hecho biológico de que tanto la piel como el sistema nervioso (que incluye lógicamente al cerebro) provienen de unos mismos tipos de células que en el embrión conformaban el llamado ectodermo. Por ese motivo, el grado de mosaicismo en la piel nos puede orientar (sólo orientar) sobre el grado de mosaicismo en el sistema nervioso.
Se suele hablar de mosaicismo celular cuando en un mismo tejido aparecen células normales y trisómicas. El porcentaje de unas y otras puede variar algo a lo largo de la vida. Y de mosaicismo tisular cuando es un único tejido el que muestra la trisomía.
CONSECUENCIAS DEL MOSAICISMO
A partir de estos datos nos movemos en una serie de incertidumbres que suelen causar a los padres mucha inquietud. Hecho el diagnóstico de mosaicismo, es lógico que deseen saber qué porcentaje de células normales hay; y como hemos visto, es posible llegar a una aproximación. En segundo lugar desean saber qué grado de afectación habrá de tener el niño en su desarrollo, tanto orgánico como mental. Y aquí nos movemos en un terreno resbaladizo, como vamos a explicar.
Consideremos el caso de los niños con trisomía 21 simple en la que todas las células de su cuerpo presentan 47 cromosomas. A pesar de que todos tienen 47 cromosomas, unos nacen con cardiopatía y otros sin ella, unos con cataratas y otros sin ellas, unos con gran hipotonía y otros con poca. Es decir, no basta que existan tres cromosomas 21 para que aparezca una determinada anomalía; hace falta que, además, los tres cromosomas 21 se expresen en un órgano concreto. Existe por tanto una gran variabilidad en la forma e intensidad con que el síndrome de Down se manifiesta en una persona concreta.
Pues bien, tomemos el caso del niño con mosaicismo. Esa variabilidad también existe e incluso con mayor intensidad. Con independencia del número de células trisómicas y normales que tenga, lo decisivo es a qué órgano u órganos afectan las trisómicas, y cuántas hay en ese órgano. Así, existen niños mosaico con muy pocos de los rasgos faciales propios del síndrome de Down y otros con igual porcentaje de mosaicismo y claros rasgos faciales. Unos con cardiopatía y otros sin ella. Y así sucede con los demás rasgos propios del fenotipo del síndrome de Down.
La pregunta crítica para los padres es: ¿Qué pasa o pasará con su desarrollo mental? ¿Qué grado de discapacidad intelectual tendrá?
MOSAICISMO Y DESARROLLO MENTAL
La discapacidad intelectual es consecuencia de las alteraciones cerebrales que se producen como consecuencia de la trisomía neuronal. Los ejemplos que hemos puesto sirven también para las células del cerebro. Con la diferencia de que el cerebro está compuesto por muchísimas más células que cualquier otro órgano y mucho más variadas. También en la trisomía 21 simple se dan casos de personas con síndrome de Down con capacidades mentales muy diferentes (al margen de la acción educativa). Lo mismo sucederá en la trisomía mosaico. Pero a ello hay que añadir la variabilidad en el porcentaje de células normales y trisómicas que pueda haber en el cerebro. Por consiguiente, todo dependerá del número de las primeras neuronas formadas durante el desarrollo embrionario y fetal que poseyeran 47 cromosomas, del número final existente, y de la función concreta que realicen. Por consiguiente, tras el diagnóstico inicial de trisomía mosaico y de haber realizado el análisis porcentual de células trisómicas, podemos hacer una presunción de que cuanto menos porcentaje haya, más probable es que el desarrollo mental sea mejor.
Son muy escasos los trabajos en que se ha evaluado y comparado de manera sistemática la calidad mental de niños con síndrome de Down mosaico frente a niños con síndrome de Down con trisomía simple. Hay uno muy citado, publicado en 1991 por K. Fishler y R. Koch (Am. J. Ment. Retard. 1991; 96: 345-351), en el que los autores compararon las adquisiciones mentales, medidas por una batería de diversos tests psicométricos, en dos grupos de tamaño y características similares, de 30 personas cada uno. En el primero todas tenían trisomía 21 simple, en el segundo tenían mosaicismo valorado por el estudio de los cromosomas en al menos 100 linfocitos. El análisis de sus resultados permite hacer las siguientes consideraciones:
En conjunto, el Coeficiente Intelectual (CI) en el grupo mosaico fue superior al del grupo con trisomía simple; y no sólo como media porque 11 personas en el grupo mosaico tenían un CI superior a 70, frente a sólo 5 del grupo trisómico.
Los casos de CI más altos estaban en el grupo mosaico. Los autores afirman, además, que la capacidad verbal del grupo mosaico era en general superior a la del grupo trisómico, así como su capacidad viso-perceptiva en tareas que requerían papel y lápiz.
Existen casos de relativamente escaso mosaicismo, a juzgar por el porcentaje de linfocitos normales, y sin embargo un CI muy bajo (casos 5, 11, 18, 20 y 29); casos de porcentajes de células normales bajo y CI moderado-alto (casos 23, 26 y 27).
A la vista de todo lo expuesto, consideramos que la actitud de los padres ante un hijo diagnosticado de síndrome de Down mosaico ha de ser la de expectación activa. Sea cual sea el diagnóstico y porcentaje de células normales, todo lo que el niño consiga depende directamente del esfuerzo que los padres pongan. Es decir, pueden alimentar la esperanza de que su hijo tenga poca afectación cerebral, y eso es bueno; pero eso no les exime de trabajar intensamente. Dada la imposibilidad de analizar directamente la trisomía en las neuronas, la conjetura es que si el niño posee, en efecto, un buen desarrollo cerebral, el trabajo de los padres se notará y los avances serán evidentes y se irán aproximando a grados altos de desarrollo. Y si la afectación cerebral es más intensa, también el trabajo de los padre será fundamental como ocurre con cualquier niño con síndrome de Down.
Las actuaciones e intervenciones serán las mismas: la intervención temprana, la intervención de la comunicación y del lenguaje, la enseñanza de la lectura, el desarrollo motor grueso y motor fino, etc. Sólo los resultados a lo largo del tiempo nos irán diciendo cómo se va desarrollando el niño e irán marcando las expectativas.
CUESTIONES ESPECIALES
Hay situaciones que pueden perturbar a los padres.
¿Qué pasa cuando el cariotipo es de síndrome de Down mosaico pero los signos externos no lo muestran claramente, de modo que incluso puede parecer normal si uno no se fija de forma especial? Podría parecer una discapacidad intelectual sin que pareciera síndrome de Down. Las consecuencias prácticas no han de diferir de lo que habría de hacerse en cualquier discapacidad intelectual, que dependerá del grado de afectación intelectual que tenga el niño. Pero resulta difícil para los padres situarse.
Una madre escribía: Los meses fueron pasando y aquel pronóstico no se fue cumpliendo…la niña creció más que un niño sin trisomía, su lengua jamás estuvo fuera, nunca fue hipotónica, caminó al año y pocos meses, es sana, hace rato largo se alimenta sola, corre, sube y baja escaleras…María tiene poco de síndrome de Down… y con esta realidad, nosotros los padres, ¿dónde la ubicamos y dónde nos ubicamos nosotros?
¿Y qué pasa cuando el desarrollo intelectual es elevado, pero el niño tiene claros signos externos de síndrome de Down, como si eso le marcara para siempre? Es lógico que los padres se preocupen. El apoyo que el niño necesitará será grande, desde muchos puntos de vista. Pero el caso no difiere del que ocurre con los niños que, teniendo trisomía completa, alcanzan un grado elevado de desarrollo intelectual.
Con frecuencia, los padres desean contactar con otros en la misma situación, guiarse por su experiencia, pese a las diferencias que existen de un individuo a otro. Existe una asociación internacional en donde se puede recibir información y apoyos. Es la siguiente:
International Mosaic Down Syndrome Association (IMDSA)»
P.O. Box 1052
Franklin, TX 77856
USA
C/o Dave Wilt
2976 Milky Way Road
Dover, PA 17315
USA
El 21 fue el segundo cromosoma humano secuenciado en el Proyecto Genoma en el año 2000. Su relevancia no está tanto en el número de genes que contiene (200-300), menos que el 22 (del mismo tamaño), sino en las funciones que median.
Sin embargo, está por demostrar si las anomalías que se observan en el síndrome de Down se deben exclusivamente a la alteración total o parcial del cromosoma 21, o si hay más información genética implicada (Letourneau et al., 2014). Se ha demostrado experimentalmente que en el embrión con síndrome de Down la actividad de los genes está alterada en todos los cromosomas y no sólo en el 21.
Generalmente, el síndrome es detectado durante el embarazo con pruebas de cribado realizadas en el primer trimestre de gestación. Si las pruebas indican un riesgo alto de afectación, existen antecedentes genéticos o la edad de la madre sobrepasa los 35 años, se confirma el diagnóstico a través de amniocentesis, muestra de vellosidades coriónicas o muestra de sangre umbilical percutánea.
En cuanto a su pregunta puedo decirle que no existe un perfil neuropsicológico «típico» del Síndrome de Down. Este síndrome presenta una variabilidad tan amplia que es prácticamente imposible describir unas características comunes a todos los casos. Sí que parece haberse reconocido, por algunas investigaciones experimentales, que los casos asociados a trisomía por mosaicismo presentan, en algunos casos, síntomas más leves de afectación en ciertos individuos.
«En cuanto a su pregunta puedo decirle que no existe un perfil neuropsicológico «típico» del Síndrome de Down. Este síndrome presenta una variabilidad tan amplia que es prácticamente imposible describir unas características comunes a todos los casos. Sí que parece haberse reconocido, por algunas investigaciones experimentales, que los casos asociados a trisomía por mosaicismo presentan, en algunos casos, síntomas más leves de afectación en ciertos individuos». Docente Maria Angeles Perez
El síndrome de Down es la cromosomopatía más frecuente de la especie humana y uno de los tipos de discapacidad intelectual más estudiados. Son pocos los rasgos característicos que se manifiestan: dismorfología facial, cerebro más pequeño e hipocelular, histopatología de la enfermedad de Alzheimer a partir de la cuarta década de vida, disfunción cognitiva e hipotonía muscular.
Como consecuencia de la alteración cromosómica, se producen anormalidades estructurales y funcionales en el SNC, que tienen como resultado diversos tipos y grados de disfunción cognitiva. Desde su primera descripcio?n, se ha asociado el si?ndrome de Down con una discapacidad intelectual considerada de moderada a grave. Por otra parte, los avances en medicina han conseguido tratar los problemas de salud de esta población (cardiopatías, malformaciones digestivas, alteraciones endocrinas, etc.) e incrementar las expectativas de vida. Pese a la homogeneidad etiológica y a la presencia de rasgos comunes, las personas con síndrome de Down son muy diferentes entre sí, y esta variabilidad se pone de manifiesto en muchas facetas, incluyendo el desarrollo neuropsicológico.
«En el síndrome de Down hay una diversidad enorme en cuanto a su sintomatología y concretamente, en la discapacidad intelectual la gran mayoría de afectados muestran una variabilidad que suele ser de muy leve a moderada. Solamente es grave o muy grave en una pequeña proporción de los casos de personas con síndrome de Down, por lo que lo más correcto sería decir que la discapacidad intelectual varía de leve a moderada». Docente Maria Angeles Perez
Recuadro 9-2. Neuropsicología y hallazgos neuroanatómicos en el síndrome de Down
En estudios post mortem se constataba la reducción del volumen de regiones como el cerebelo, el tronco cerebral, la corteza temporal, el hipocampo y la corteza frontal, entre otras (Fernández-Alcaraz y Carvajal-Molina, 2014). También establecieron, a una edad temprana, hallazgos neuropatológicos similares a la enfermedad de Alzheimer, como ovillos neurofibrilares, placas seniles y respuestas cerebrales inflamatorias. Dichas alteraciones aumentan con la edad, por lo que, a medida que envejecen, se incrementa el riesgo de presentar demencia, estimada desde un 15% a los 45 años hasta el 75% después de los 65 (Coppus et al., 2006). El diagnóstico de demencia puede resultar problemático debido a la gran variabilidad intraindividual en el funcionamiento cognitivo y a la dificultad para identificar signos precoces de deterioro en personas que exhiben un déficit intelectual previo, ya que no son adecuadas las pruebas neuropsicológicas que se utilizan para la población general con frecuente «efecto suelo».
La neuroimagen estructural muestra diferencias volumétricas. La inconsistencia entre investigaciones en relación con el tamaño de algunas estructuras se explica por la dificultad en disociar las anomalías cerebrales propias del síndrome de los cambios degenerativos debido al envejecimiento acelerado que sufre el cerebro en esta población. Dichas diferencias se han empezado a correlacionar con las alteraciones en el perfil neuropsicológico característico de estos niños. Así, la reducción en el tamaño del cerebelo se asocia a déficits motores y en lenguaje; la mayor preservación de estructuras parietales y occipitales con el mejor desempeño en funciones visoespaciales; las anomalías en el hipocampo con los problemas en la memoria episódica, y la reducción en la corteza frontal y la circunvolución del cíngulo con alteraciones en la atención y las funciones ejecutivas (Carducci et al. 2013).
A nivel microscópico se observan déficits en la arborización de las dendritas y en la estructura laminar de la corteza, reducción de sinapsis y retraso en la mielinización. Todas estas alteraciones repercutirán en la red de conexiones entre distintas zonas cerebrales.
Investigaciones con RMf han mostrado un patrón de actividad cerebral diferente incluso en tareas en las que tienen un desempeño similar al de su grupo de edad (Jacola et al., 2011), lo que refuerza la hipótesis de que las alteraciones en el desarrollo pueden configurar un cerebro diferente no sólo en la estructura, sino también en su funcionamiento.
Recuadro 9-2. El desarrollo temprano de los niños con síndrome de Down
Los nin?os con si?ndrome de Down muestran, en relacio?n con aquellos con un desarrollo ti?pico o con otros tipos de discapacidad intelectual, un fenotipa distintivo compuesto por un patro?n caracteri?stico de de?ficits y fortalezas (Fidler, 2005):
Retraso en el desarrollo psicomotor. Los hitos motores básicos se adquieren en el mismo orden, pero en edades posteriores. El desarrollo de la motricidad estará influido por características propias del síndrome (hipotonía muscular, laxitud de ligamentos, reducción de la fuerza y el tamaño de las extremidades superiores e inferiores, que son más cortas). Es frecuente la torpeza motora gruesa y fina, la lentitud, la deficiente coordinación oculomanual y dinámica y las dificultades de equilibrio. Estos retrasos en el desarrollo motor y en el control postural limitan las experiencias motrices y la exploración del entorno (Candel, 2005).
Déficit en los sistemas de atención y alerta. Conducta dispersa, con poco interés por los estímulos ambientales. Son proclives a centrarse en los aspectos poco relevantes, olvidando los más significativos. Frecuente tendencia a la distracción y elevada sensibilidad a la interferencia.
Alteración en la adquisición y el desarrollo del lenguaje. Las dificultades de comunicación son ya evidentes en la fase prelingüística, desde los primeros meses de vida. No sólo existe un retraso en la aparición de la sonrisa y el contacto ocular, sino que no se utiliza ninguno de ellos como medio para iniciar y mantener una comunicación. Aunque las primeras vocalizaciones se manifiestan de forma similar a las de los niños “normales”, se van empobreciendo y las palabras aparecen muy tardíamente (Alonso Hernández, 2010). Una vez que el niño ha adquirido el lenguaje, su capacidad expresiva es inferior a su capacidad comprensiva. Saben lo que quieren decir pero les cuesta expresarlo y fracasan al tratar de transmitir sus ideas o sentimientos. Tienen dificultades articulatorias, lo que provoca dislalias de varios fonemas, como /b/, /c-k-q/, /d/, /rr/, /g/ y /j/. Suelen hablar atropelladamente, a borbotones, con pocas pausas; el volumen también puede estar alterado (por exceso o por defecto) y la voz suele ser ronca, grave, áspera, carente de timbre. A nivel morfológico presentan dificultades para formar familias de palabras, con aumentativos-diminutivos, singular-plural, masculino-femenino, así como con las concordancias entre género y número. Las frases suelen ser telegráficas con estructuras sintácticas simples. En cuanto al contenido, su vocabulario suele ser reducido o limitado a nombres y objetos del entorno cercano. Presentan dificultad para evocar palabras, así como para estructurar su vocabulario en diferentes campos semánticos. Además tienen dificultades con palabras que denotan tiempo (ayer, hoy, mañana, días de la semana), lo que repercute en la capacidad para describir o narrar sucesos en orden cronológico. Pragmáticamente se caracterizan por tener menor intención comunicativa y no suelen respetar las normas conversacionales: guardar turnos, mantener la distancia adecuada, etc. (Alonso Hernández, 2010).
Lateralización anormal. Estudios recientes refieren a estos pacientes un mayor porcentaje de casos con dominancia manual mixta e izquierda que en la población general, lo que podría estar en la base de los problemas lingüísticos que caracteriza este síndrome.
Mayor dificultad para procesar la información auditivoverbal que la visoespacial. La capacidad de procesamiento visoespacial se considera una fortaleza de su perfil cognitivo.
Alteraciones amnésicas. Su memoria a corto plazo (en especial para material auditivo verbal) es muy limitada. Suelen fracasar en la consolidación del conocimiento y en la adquisición de nuevas habilidades, siendo el proceso de aprendizaje más lento. Son capaces de retener poca información y necesitan más tiempo, por lo que la brevedad del mensaje y la repetición es fundamental para que la información se consolide. La memoria a largo plazo estaría menos afectada, aunque muestran especial dificultad para recordar información almacenada de manera consciente (memoria explícita o declarativa). Sin embargo, la memoria implícita o de actos motores suele estar intacta y ser similar a la que presentan los niños con desarrollo normal.
Déficit ejecutivo. Las capacidades de planificación, abstracción de reglas, generalización, inhibición y memoria de trabajo se encuentran alteradas. Se observa persistencia de conducta, bajo nivel de espontaneidad y resistencia a los cambios y situaciones nuevas. Son proclives a un uso de conductas de evitación e impulsivas cuando se enfrentan a retos nuevos o a aprendizajes por encima de su nivel (Candel,2005). Las dificultades en el razonamiento matemático y el cálculo también son frecuentes.
Tabla 9-3. Perfil neuropsicológico del Síndrome de Down y su correlación con estructuras cerebrales
| Proceso | Perfil neuropsicológico | Estructuras afectadas |
|---|---|---|
| Memoria | Más preservada la memoria implícita que la declarativa. Dificultad en la memoria episódica. Más preservada la memoria visual que la verbal. Déficits en la memoria de trabajo. Dificultades en la memoria a largo plazo con material verbal. | Hipocampo Lóbulo temporal Lóbulo frontal |
| Atención | Dificultades concentración. Mayor tendencia a la distracción. | Lóbulo frontal Cerebelo |
| Percepción | Déficits en habilidades visoperceptivas. Dificultades en la realización de tareas perceptomotoras. | Cerebelo y conexiones frontocerebelosas |
| Lenguaje y comunicación | Buena capacidad de imitación. Uso adecuado de gestos. Buena capacidad para adquirir vocabulario. Dificultades en morfosintaxis. Buen desempeño en la lectura. Mayores capacidades receptivas que expresivas. | Circuito perisilviano frontal |
| Emoción | Buena respuesta emocional. Puede aparecer depresión como comorbilidad. | Mejor preservación del sistema límbico que en otros síndromes |
| Funciones ejecutivas | Falta de iniciativa. Dificultades en la planificación. Problemas en el razonamiento abstracto. Déficits en la secuenciación espacio temporal. Inflexibilidad en el cambio de estrategias para solucionar nuevos problemas. | Corteza prefrontal |
RESUMEN
Juicio clínico: El síndrome de Down es la cromosomopatía más frecuente de la especie humana. Como consecuencia de la alteración cromosómica, se producen anormalidades estructurales y funcionales en el SNC, que dan como resultado diversos tipos y grados de disfunción cognitiva. La mayoría de las personas con Síndrome de Down tiene una discapacidad intelectual de leve a moderada.
- Hallazgos neuropatológicos: como alteraciones macroscópicas aparecen microcefalia y retraso en el crecimiento del cerebro, reducción en el volumen de los lóbulos frontales y en el tamaño del tronco cerebral, el hipocampo y el cerebelo. Las anomalías microscópicas se relacionan con:
- Disminución de determinados tipos de neuronas situadas en la corteza cerebral.
- Alteración en la estructura y número reducido de espinas dendríticas.
- Perfil neuropsicológico: dificultades para procesar la información auditiva (funcionan mejor en tareas que incluyen las modalidades visual-motor y visual-vocal), problemas de atención y tendencia a la conducta impulsiva. Los retrasos en desarrollo motor y control postural limitan la exploración de su entorno. Se observan déficits de comunicación (los problemas de imitación vocal y de expresión verbal agudizan las dificultades de lenguaje).
La intervención ha de adaptarse a las características de los niños en cada etapa de su vida. Conviene recurrir a los apoyos visuales y a las actividades funcionales para mejorar la adquisición de habilidades. La inclusión en un contexto escolar normalizado favorece los aprendizajes y las competencias que les permitirán alcanzar un adecuado grado de autonomía e independencia personal y social. La colaboración con la familia es indispensable. Desde el principio, un objetivo fundamental en el trabajo con los padres es conseguir unos niveles de adaptacio?n que ayuden a su hijo en la integración social.
ESQUEMA
AUTOEVALUACIÓN
En exámenes anteriores preguntaron…
| Los niños con Síndrome de Down afectados por la variante de mosaicismo mostrarán un fenotipo característico que dependerá de que haya varios órganos afectados por células con esa trisomía. |
| Habitualmente distintos grados y tipos de disfunción cognitiva. |
| El perfil neuropsicológico del Sindrome de Down se caracteriza entre otros síntomas, por una mayor capacidad receptiva del lenguaje que expresiva. Esto correlaciona con la alteración del circuito perisilviano frontal. |
| déficit en las arborizaciones dendríticas. |
| En el desarrollo temprano de los niños con síndrome de Down es frecuente que tengan una mayor torpeza motora gruesa y fina. |
REFERENCIAS
- Arnedo Montoro, M. (2018). Neuropsicología del desarrollo. Madrid: Médica Panamericana.
- Apuntes CARMEN ORTEGO
- YouTube